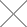Li Zhanting, Zhao Jincai. PREFACE[J]. Progress in Chemistry, 2020, 32(11): 0-0.

Qian Zhou, Na Li, Kun Li, Xiaoqi Yu. Detection of 5-Formylpyrimidines in DNA Based on Chemoselective Labeling[J]. Progress in Chemistry, 2020, 32(11): 1634-1650.
In addition to the four canonical nucleobases of A, G, T and C, hundreds of chemical modifications have been identified in genome DNA. Among them, 5-formylpyrimidines, including 5-formylcytosine(5fC) and 5-formyluracil(5fU), are naturally occurring pyrimidine-covalent-modifications and are widely distributed in mammalian cells. Emerging evidence indicates that 5fC not only serves as a key intermediate in active DNA demethylation but also carry independent epigenetic significance; while 5fU was always considered to be an oxidative DNA lesion with high genotoxicity. In order to further understand their regulatory functions, it is necessary to develop accurate, sensitive and facile methods for qualitative, quantitative, and localized detection of 5-formylpyrimidines in the whole genome. In this review, we summarized the recent progress in detecting 5-formylpyrimidines from the perspective of selective chemical labeling.
1 Introduction
1.1 DNA methylation and demethylation
1.2 5-Formylcytosine and 5-formyluracil
1.3 Detection methods of 5-formylpyrimidines
2 Detection of 5-formyluracil based on chemoselective modification
2.1 Fluorescent labelling of 5-formyluracil
2.2 Enrichment and sequencing of 5-formyluracil
3 Detection of 5-formylcytosine based on chemoselective modification
3.1 Fluorescent labelling of 5-formylcytosine
3.2 Enrichment and sequencing of 5-formylcytosine
4 Conclusion and outlook
Hui-Juan Wang, Yu Liu. Molecular Binding and Assembly of Sulfonated Crown Ethers[J]. Progress in Chemistry, 2020, 32(11): 1651-1664.
Crown ethers, as the first generation macrocycle, with flexible cavity, are widely used to construct supramolecular assemblies, due to their complexation of metal ions and organic cations. Sulfonated crown ether is a kind of anionic crown ether derivative with good water solubility, compared with crown ether, it possess more binding sites, stronger binding ability and better guest selectivity with metal ions and organic cations. This review introduces the research progress of sulfonated crown ether from the synthesis of sulfonated crown ethers, the complexation of alkali metal ions, lanthanide metals, and the assembly of organic cationic guests. Then we comprehensively analyse the binding modes and driving forces of sulfonated crown ethers from the perspectives of thermodynamics and crystal structures. Finally, we discuss the opportunities and challenges of the development of molecular binding and assembly of sulfonated crown ethers, and prospected the application of sulfonated crown ethers.
1 Introduction
2 The sulfonation method of crown ether
3 The binding and assembly of sulfonated crown ether to metal ions
4 The binding and assembly of sulfonated crown ether to organic cations
4.1 The binding and assembly of bis-sulfonated crown ether to organic cations
4.2 The binding and assembly of tetra-sulfonated bis(m-phenylene) crown ether to organic cations
4.2.1 The binding and assembly of tetra-sulfonated dibenzo crown ether to organic cations
4.2.2 The binding and assembly of tetra-sulfonated dinaphtho crown ether to organic cations
5 Conclusion
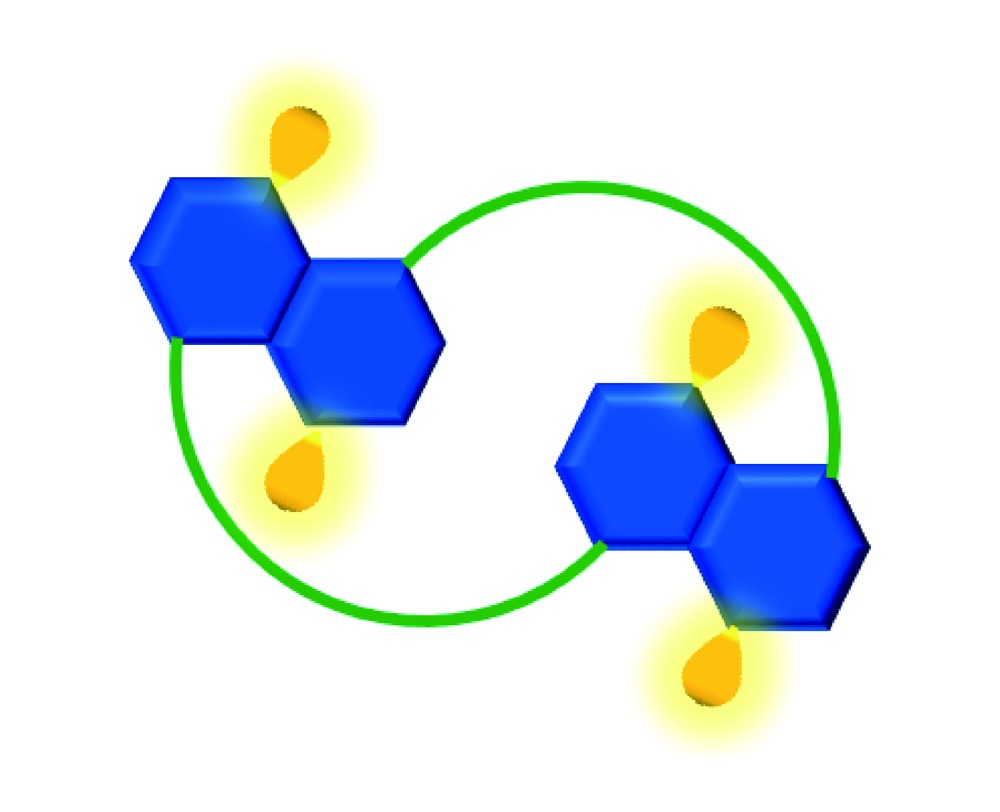
Dan-Wei Zhang, Hui Wang, Zhan-Ting Li. Macromolecular and Supramolecular Helical Tubes: Synthesis and Functions[J]. Progress in Chemistry, 2020, 32(11): 1665-1679.
It has been established that hydrophobicity, hydrogen bonding, electrostatic attraction, halogen bonding and coordination all can be utilized to stabilize the folding and/or helicity of aromatic macromolecules and supramolecular systems. The resulting aromatic helical tubes possess defined diameters and tunable depth and can be used as synthetic receptors for recognizing or encapsulating a variety of guest species, achieving chirality induction and transfer, promoting organic transformations, and as artificial channels for transmembrane transport. This review highlights the advance in the construction of such family of macromolecular and supramolecular tubes from aromatic segments and their important functions. In the first section, we introduce the background of the formation of tubular structures from different strategies and the features of the supramolecular and macromolecular approaches. We then highlight the formation of molecular tubes from various oligomeric molecules with aromatic amide, hydrazide, triazole and ethyne repeat segments. In the following section, we describe the use of polymeric backbones with the above repeat segments. The self-assembly strategy for the formation of supramolecular tubes is then summarized in another section. In particular, the utility of hydrogen bonding, halogen bonding and coordination interactions has been described. When available, the functions of the tubular structures are briefly presented. In the last section, we discuss the synthetic challenges for the formation of long macromolecular tubes and the new potential applications of this family of structurally unique structures.
1 Introduction
2 Aromatic oligomeric tubes
2.1 Aromatic amide and hydrazide backbones
2.2 Aromatic triazole backbones
2.3 Aromatic ethyne backbones
3 Aromatic polymeric tubes
3.1 Aromatic amide backbones and analogues
3.2 Aromatic triazole and oxadiazole backbones
3.3 Aromatic ethyne backbones
4 Aromatic supramolecular tubes
5 Conclusion and outlook
Xin Lin, Fanfu Guan, Jialin Wen, Pan-Lin Shao, Xumu Zhang. Synthesis of Chiral Tridentate Ligands with a Ferrocene Framework and Their Applications in Ir-Catalyzed Asymmetric Hydrogenation[J]. Progress in Chemistry, 2020, 32(11): 1680-1696.
In this review, the synthesis and application of a series of tridentate ligands, such as f-amphox, f-ampha, f-amphol and f-amphamide, bearing ferrocene skeleton are introduced. The corresponding iridium complex based on these chiral P-N ligands can catalyze(functionalized) ketones with high TON(up to 1 000 000), excellent conversion(>99%) and enantioselectivity(>99% ee). The application of the f-series ligands has successfully realized the asymmetric synthesis of many chiral pharmaceutical intermediates such as Dinopramine, Phenylephrine and Salbutamol. These approaches are more efficient, generating less by-products and lower industrial emissions compared with the traditional routes.
1 Introduction
2 History of tridentate ferrocene ligands
2.1 The development of tridentate ligands
2.2 Special properties of ferrocene skeleton
2.3 Representative ligands containing ferrocene framework
3 f-amphox
3.1 The synthesis of f-amphox
3.2 Application of f-amphox in asymmetric hydrogenation
4 f-amphol
4.1 The synthesis of f-amphol
4.2 Application of f-amphol in asymmetric hydrogenation
5 f-ampha
5.1 The synthesis of f-ampha
5.2 Application of f-amphol in asymmetric hydrogenation
6 f-amphamide
6.1 The synthesis of f-amphamide
6.2 Application of f-amphamide in asymmetric hydrogenation
7 New mechanism insight about asymmetric hydrogenation
7.1 Hydrogenation mechanism of N—H structure
7.2 The chiral sources of f series
7.3 Transition state model of asymmetric hydrogenation
8 Conclusion and outlook

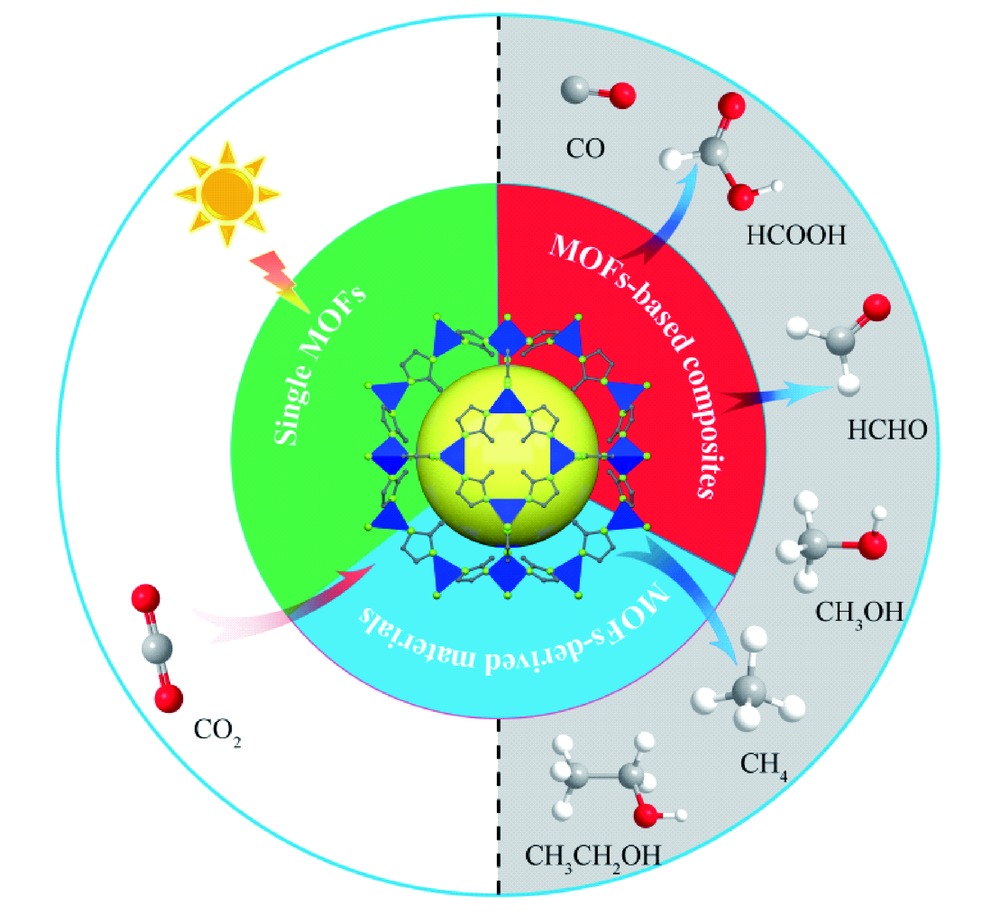
Xiao Feng, Yanwei Ren, Huanfeng Jiang. Application of Metal-Organic Framework Materials in the Photocatalytic Carbon Dioxide Reduction[J]. Progress in Chemistry, 2020, 32(11): 1697-1709.
The effective use of CO2 has become a research hotspot worldwide, whose excessive emission led to increasingly serious global environmental problems. Compared with high energy-consuming CO2 capture and storage(CCS) technology, the photocatalytic conversion of CO2 into a valuable energy fuel is an effective way to solve energy and environmental problems. Among them, the development of a photocatalyst with efficient catalytic performance under visible light is the key to this process. Currently, there are still many shortcomings in photoreduction CO2 catalysts, such as weak visible light response ability, high recombination rate of photo-generated electron-hole pairs, low CO2 adsorption capacity, poor product selectivity, and hydrogen-evolution competition in an aqueous environment. Metal-organic frameworks(MOFs), with adjustable porous structures, fast electron migration rate, large CO2 adsorption capacity, etc., are a unique class of porous crystalline materials composed of metal ions/clusters and organic ligands, which have broad application potential in CO2 photocatalytic reduction. The existing methods improving the catalytic performance of MOFs-based catalysts is mainly to enhance the absorption of visible light by functional modification, formation of composites with other functional materials and so on. This review mainly analyzes and discusses the recent advances of MOFs-based photoreduction CO2 catalysts(single MOFs, MOFs-based composites and MOFs-derived materials), and predicts future development trends and prospects of MOFs-based materials in photocatalytic reduction of CO2.
1 Introduction
2 Single MOFs
2.1 Organic ligands as photosensitizer
2.2 Metalloligands as photosensitizer
3 MOFs-based composites
3.1 MOFs/semiconductor composites
3.2 MOFs/perovskite quantum dot composites
3.3 MOFs/noble metal nanoparticle composites
3.4 MOFs/enzyme composites
4 MOFs-derived materials
5 Conclusion and outlook
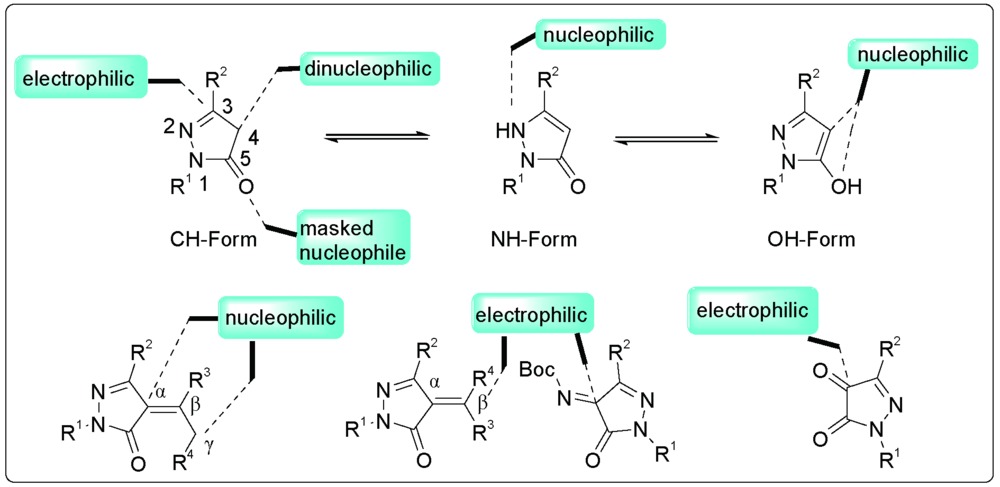
Li Luyao, Xu Xinyao, Zhu Bo, Xu Xinyao. Application of Pyrazolone Compounds in Catalytic Asymmetric Reactions[J]. Progress in Chemistry, 2020, 32(11): 1710-1728.
Pyrazolin-5-one compounds play an important role in bioactive compounds and have attracted extensive attention. Pyrazolin-5-one and its common derivatives have many reaction sites and can participate in a variety of asymmetric reactions, such as asymmetric addition reactions, asymmetric annulation reactions and other asymmetric reactions. In this context, we summarize the recent progress in asymmetric reactions of pyrazolin-5-one and its common derivatives. The main reaction types involving the pyrazolin-5-one compounds are classified, and their main reaction sites are expounded. Finally, the challenges and development of pyrazolone derivatives are summarized, and the future development direction is prospected.
1 Introduction
2 The synthesis of pyrazolin-5-one derivatives
3 Asymmetric addition reactions
3.1 Asymmetric Michael addition
3.2 Asymmetric Mannich reaction
3.3 Other asymmetric addition reaction
4 Asymmetric annulation reactions
5 Other asymmetric reactions
6 Conclusion and outlook
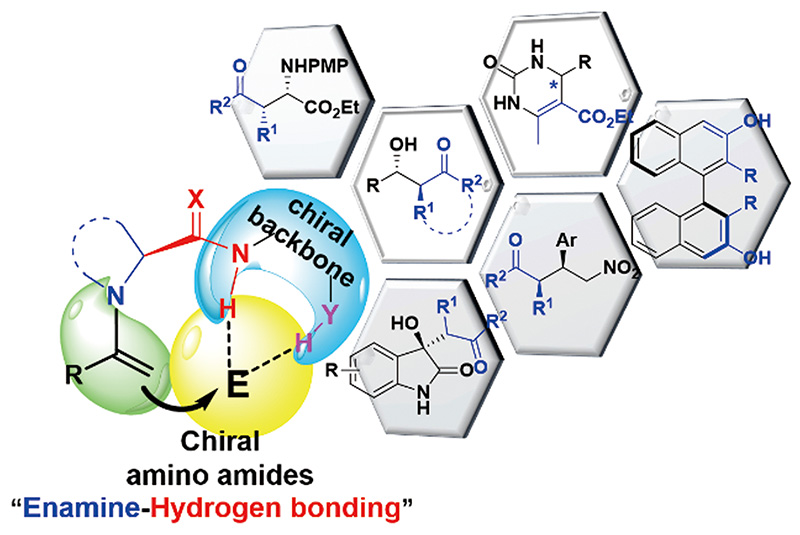
Jie Yu, Liu-Zhu Gong. Discovery and Typical Advances of Chiral Amino Amide Catalysts[J]. Progress in Chemistry, 2020, 32(11): 1729-1744.
Since L-prolinamide was revealed to have high capacity to catalyze asymmetric aldol reaction, great advances have been made on the design of chiral amino amide catalysts and their applications in asymmetric catalysis. In particular, the “enamine-double hydrogen-bonding activation mode” has turned out to be a general concept for the proliferation of structurally diverse range of organocatalysts. This review mainly describes asymmetric reactions catalyzed by chiral amino amides containing single hydrogen-bonding donor, double hydrogen-bonding donors and multiple hydrogen-bonding donors, including enantioselective direct aldol reaction, Mannich reaction, Michael addition reaction, cycloaddition reaction, tandem cyclization reaction, Biginelli reaction and others.
1 Introduction
2 Chiral amino amide catalysts without hydrogen-bonding donor
3 Chiral amino amide catalysts with single hydrogen-bonding donor
3.1 Asymmetric direct aldol reaction
3.2 Asymmetric Mannich reaction
3.3 Asymmetric Michael addition
3.4 Asymmetric cascade cyclization
3.5 Asymmetric cycloaddition
3.6 Miscellaneous reactions
4 Chiral amino amide catalysts with double hydrogen-bonding donors
4.1 Asymmetric direct aldol reaction
4.2 Asymmetric Michael addition
4.3 Miscellaneous reactions
5 Chiral amino amide catalysts with multiple hydrogen-bonding donors
6 Conclusion and outlook
Chenghao Zhu, Junliang Zhang. Palladium Catalyzed Heck-Type Reaction of Organic Halides and Alkyl-Alkynes[J]. Progress in Chemistry, 2020, 32(11): 1745-1752.
As an important unit of molecular structure and organic building blocks, the development of new synthetic methods of allene has received much attention. The Heck and related cascade reaction of alkenes is arguably one of the most synthetically versatile method. However, the Heck reaction of alkyl-alkynes leading to allenes has lagged behind due to the energetically unfavored β-hydride elimination of vinyl palladium species. Several competitive reactions such as protonation, carbohalogenation and cascade reaction exist. Moreover, the isomerization of allene and the regioselectivity of unsymmetrical alkyl-alkynes also limited this research progress. To effectively promote β-hydride elimination of vinyl palladium species, several commonly-used strategies are the increasing reaction temperature, introduction of ortho-substituent to aryl halides and the development of ligand. In this mini-review, Heck and related reaction of alkyl-alkynes including β-hydride elimination reactions of vinyl palladium species, protonation reaction, carbohalogenation reaction, and cascade reaction are emphasized. Finally, the major limitation and an outlook this type of reaction are provided.
Contents
1 Introduction
2 Heck-type reaction pathway of alkyl-alkynes
3 Heck reaction of alkyl-alkynes
4 Other competitive reactions
4.1 Carbohalogenation process
4.2 Domino-Heck cyclization process
4.3 Activation process of aryl C-H
5 β-hydride elimination reactions
6 Conclusion and outlook
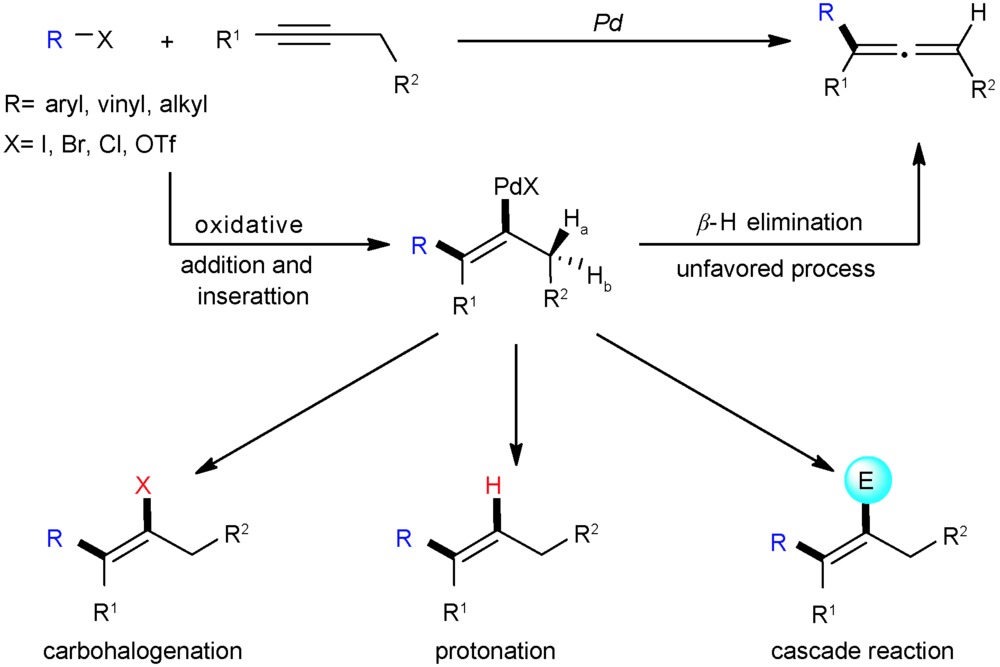
Ruipu Zhang, Runze Zhang, Sanzhong Luo. Bio-Inspired ortho-Quinone Catalysis[J]. Progress in Chemistry, 2020, 32(11): 1753-1765.
Quinoproteins are an important type of redox enzymes for the oxidative metabolism of alcohols and amines, which employ ortho-quinones as the cofactors. On the basis of catalytic principles and strategies of quino-enzymes, a number of molecular quinone catalysts have been developed during the past two decades. With copper amine oxidases(CuAOs) as a blueprint, small molecular ortho-quinone catalysts have been developed for amine oxidation. These catalysts could mimic the catalytic performance of CuAOs, and expand the substrate scope to α-branched primary amines, secondary amines and tertiary amines. Recent studies have also uncovered a new type of quinoproteins, methanol dehydrogenase(MDH), utilizing rare earth elements as the active metal. In this review, we summarize the major types of quinoproteins, the development of their mimics ortho-quinone catalyst as well as perspective on the future development of bio-inspired ortho-quinone catalysis.
1 Introduction
2 Quinoprotein
2.1 Alcohol dehydrogenase
2.2 Copper amine oxidase
3 Bio-inspired ortho-quinone catalysis
3.1 Amine oxidation
3.2 Alcohol oxidation
4 Conclusion and outlook
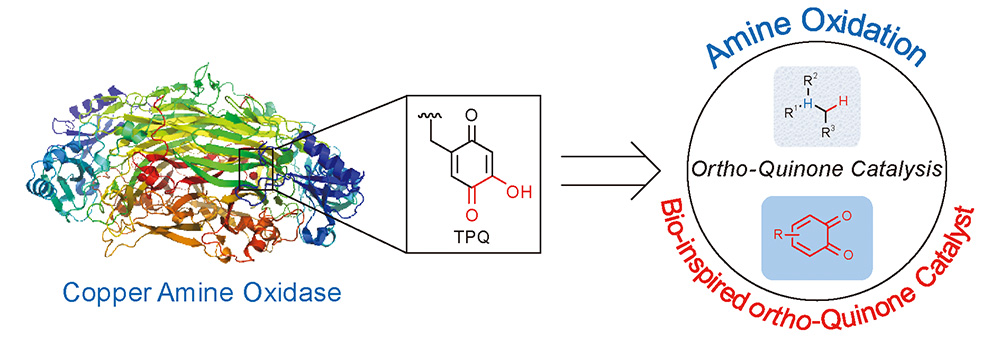
Huina Zou, Shoufei Zhu. Progresses of 1,10-Phenanthroline Type Ligands in Fe/Co/Ni Catalysis[J]. Progress in Chemistry, 2020, 32(11): 1766-1803.
1,10-Phenanthroline and its derivatives, classic bidentate N-donor ligands, can form stable complexes with a variety of transition metals and have been widely used as catalysts in various organic reactions. Ferritic elements(iron, cobalt, nickel) have the advantages of high natural abundance, low cost, low toxicity and unique catalytic performance. The complexes of 1,10-phenanthroline type ligands and ferritic elements are ideal alternative catalysts. In recent years, 1,10-phenanthroline type ligands have been widely used in Fe/Co/Ni-catalyzed organic reactions,especially in cross-coupling reactions, addition reactions,and redox reactions,showing unique ligand effects. More and more studies revealed that the rigid aromatic structure of 1,10-phenanthroline plays an important role in improving the stability of the catalyst, and the substituents of 1,10-phenanthroline have a significant impact on the activity and selectivity of corresponding catalyst. More interestingly, a few recent studies disclosed that 1,10-phenanthroline ligands might change the spin state and three-dimensional electronic structure of the Fe/Co/Ni catalysts, which accounts for their unique reactivity as well as selectivity.Although with the above-mentioned progresses, there are still several important challenges in this field, including the poor structural diversity of 1,10-phenanthroline type ligands and poor understanding of the electron effect of 1,10-phenanthroline ligands to corresponding metal catalysts.In this review, we summarized the applications of 1,10-phenanthroline ligands in Fe/Co/Ni-catalyzed organic reactions, and gave an outlook of this promising field.
1 Introduction
2 Application of 1,10-phenanthroline type ligands in Fe-catalyzed reactions
2.1 Coupling reactions
2.2 Oxidation reactions
2.3 Reduction reactions
2.4 Addition reactions
2.5 Other reactions
3 Application of 1,10-phenanthroline type ligands in Co-catalyzed reactions
3.1 Addition reactions
3.2 Cycloaddition reactions
3.3 C—H functionalization reactions
3.4 Carboxylation reactions
3.5 Coupling reactions
3.6 Other reactions
4 Application of 1,10-phenanthroline type ligands in Ni-catalyzed reactions
4.1 Cross-coupling reactions
4.2 Reductive coupling reactions
4.3 Oxidation reactions
4.4 Hydrogen-borrowing reactions
4.5 Decarboxylative coupling reactions
4.6 Addition reactions
4.7 Oxidative reactions
5 Conclusion and outlook
Hanyu Zhang, Meng Liu, Xia Wu, Miao Liu, Decai Xiong, Xinshan Ye. Photo-/Electro-Driven Carbohydrate-Based Reactions[J]. Progress in Chemistry, 2020, 32(11): 1804-1823.
Carbohydrates are the most abundant organic compounds in nature, which play essential roles in the fields of medicine, materials, energy and environment. Due to the specific characteristic of high polarity, sophisticated structure and microheterogeneity, it is particularly difficult to separate, purify and synthesize carbohydrates. At present, the acquirement of carbohydrate compounds with homogeneous and diverse structures and in high purity mainly depends on chemical synthesis. However, the effective synthesis of carbohydrates has become a bottleneck of restricting the progress of glycoscience. The development of new methods and strategies for the synthesis of carbohydrates is the core topic in carbohydrate chemistry. The reaction driven by photon/electron energy usually takes place under mild conditions, which meets the requirements of green chemistry and sustainable development, and is also one of the research frontiers in modern organic synthesis. Nowadays, with the development of photo-/electro-synthetic technology, many achievements have been made in carbohydrate-based reactions driven by photon/electron energy. In view of reaction type, mechanism and status, this review will systematically summarize the latest advances on photo-/electro-driven reactions in the synthesis of carbohydrates. This review will also analyze the current challenges and new opportunities of carbohydrate-based reactions driven by photon/electron energy.
1 Introduction
2 Photo-/electro-driven glycosylation
2.1 Thioglycosides as the donors
2.2 Selenoglycosides as the donors
2.3 Telluroglycosides as the donors
2.4 O-glycosides as the donors
2.5 Glycosyl trichloroacetimidates as the donors
2.6 Glycosyl halides as the donors
2.7 Glycals as the donors
3 Photo-driven modifications of carbohydrates
3.1 Photo-driven reductive defunctionalization
3.2 Photo-driven halogenation
3.3 Photo-driven functionalization on the anomeric O-/S-atoms
3.4 Photo-driven decarboxylation
3.5 Photo-driven isomerization
4 Conclusion and outlook

Yudong Yang, Jingsong You. Chelation-Assisted C—H/C—H Oxidative Cross-Coupling/Cyclization for the Construction of Fused(Hetero)aromatics[J]. Progress in Chemistry, 2020, 32(11): 1824-1834.
The synthesis of fused heteroaromatics is an important research area in organic chemistry and it has significant implications on the investigations of organic optoelectronic materials and devices. C—H bond is one of the most ubiquitous chemical bonds in organic compounds. The direct construction of fused heteroaromatics via the C—H bond cleavage and direct functionalization features concision and high efficiency, which could solve the limitations on the substrate preparation and product structural diversity in conventional synthetic methods. In this account, we review our works on the construction of fused heteroaromatics based on chelation-assisted transition metal-catalyzed oxidative C—H/C—H cross-coupling between(hetero)arenes/intramolecular cyclization during the past six years, including a comprehensive discussion of the features, advantages and reaction mechanisms of these reactions, and the applications of these strategies in the exploration of novel organic optoelectronic materials. Finally, the existing problems and development prospect of this area are elaborated.
1 Introduction
2 Construction of ladder-type fused heteroaromatic frameworks
2.1 Construction of pyrrole-fused ladder-type heteroaromatic frameworks
2.2 Construction of furan-fused ladder-type heteroaromatic frameworks
3 Construction of fused benzo[de]thiochromenes
4 Construction of fluorenone- and coumarin-type frameworks
5 Construction of(hetero)aryl-fused sultams and thiophene 5,5-dioxides
6 Construction of phenanthridine- and phenanthrone-type frameworks
6.1 Construction of phenanthridine-type frameworks
6.2 Construction of phenanthrone-type frameworks
7 Construction of cationic fused heteroaromatic frameworks
8 Conclusion and outlook

Miao Qian, Yang Daiyue. From Polycyclic Arenes Containing Eight-Membered Rings to Negatively Curved Nanocarbons: Progress and Outlook[J]. Progress in Chemistry, 2020, 32(11): 1835-1845.
Carbon allotropes comprised exclusively of sp2 hybridized carbon atoms present flat or curved surface, whose overall geometric feature is reflected by its curvature. Consisting of six-membered rings exclusively, graphene has zero curvature. Five-membered rings induce positive curvature as displayed in fullerenes; while seven- or eight-membered rings in a hexagonal lattice of carbon induce negative curvature as displayed in a saddle-shaped surface. Negatively curved nanocarbons of three-dimensional periodic structures are named as Mackay crystal or carbon schwarzites, which are a long-sought target in carbon nanoscience, but have not been synthesized unambiguously yet. A bottom-up approach to negatively curved nanocarbons involves synthesis of negatively curved polycyclic arenes, which can then be used as templates or monomers in synthesis of larger nanocarbons. Negatively curved polycyclic arenes, which can be designed and synthesized by introduction of seven or eight-membered rings into polycyclic aromatic frameworks, exhibit structural characteristics and properties that are not available to planar polycyclic arenes. Taking the polycyclic arenes containing eight-membered rings as examples, this review article describes research in the design, synthesis, stereochemical dynamics, and other characteristics of negatively curved polycyclic arenes, and provides an outlook for new directions in the research of negatively curved nanocarbons.
1 Introduction: negatively curved nanocarbons
2 Building blocks containing eight-membered rings
3 Design and synthesis of negatively curved polycyclic arenes
4 Stereochemistry and other characteristics of negatively curved polycyclic arenes
5 Conclusion and outlook
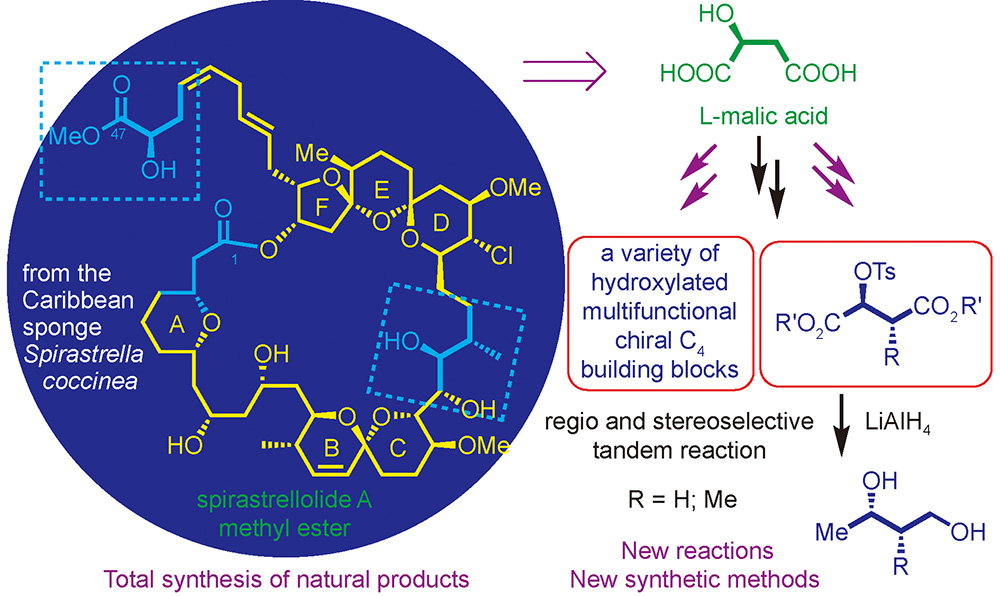
Luo Shipeng, Huang Peiqiang. Malic acid——A Versatile Chiral Building Block in the Enantioselective Total Synthesis of Natural Products and in Synthetic Methodologies[J]. Progress in Chemistry, 2020, 32(11): 1846-1868.
Building block-approach is one of the basic strategies used by nature in biogenetic synthesis. Naturally occurring L-α-amino acids, L-α-hydroxy acids, D-mono-sugars, and terpenes, being cheap and easily available in enantiopure forms, have found widespread applications enantioselective synthesis as chiral building blocks. L-Malic acid, an L-α-hydroxy diacid, is a readily available and cheap natural chiron, D-malic acid is also commercially available although it is more expensive. With all of the four carbon atoms transformable or functionalizable, malic acid serves as a versatile C4 building block in organic synthesis for enantioselective synthesis. In this review, the progress in the applications of malic acid in organic synthesis is summarized. Firstly, the transformations of malic acid into a variety of advanced chiral C4 building blocks are compiled. Secondly, selected examples on the applications of these versatile intermediates in the total synthesis of complex natural products in recent years are presented. Besides, the developments of malic acid-based new synthetic methodologies including the contribution from one of the authors’ laboratory are highlighted. An outlook for future development of malic acid-based synthetic strategy is provided.
1 Introduction
2 Classical transformations of L-malic acid
3 Application of L-malic acid in the enantioselective total syntheses of natural products
3.1 Total Syntheses based on 1b-related building blocks
3.2 Total Syntheses based on 1i-related building blocks
3.3 Total Syntheses based on 1m-related building blocks
3.4 Total Syntheses based on 1r-related building blocks
4 New synthetic methodologies based on malic acid
5 Malimide-based asymmetric synthetic methodologies
6 Conclusion and outlook