Ran Jia, Jian Wang, Wenfu Yan. From Condensed Matter Physics to Condensed Matter Chemistry[J]. Progress in Chemistry, 2026, 38(1): 1-22.
Since the widespread acceptance between the 1960s and 1970s, condensed matter physics has undergone rapid development. Condensed matter physics primarily investigates the geometric and electronic structures of solid and liquid substances, as well as the resulting microscopic and macroscopic physical phenomena such as sound, light, electricity, magnetism, heat, etc. Meanwhile, the field of chemistry has also evolved significantly, especially in the last two decades, with advancements in theoretical chemistry and chemical characterization techniques. Researchers have gradually come to realize that chemical reactions are not merely straightforward transformations from reactants to products. The structural hierarchy of the reaction system plays a crucial role in the progression of chemical reactions. There has been a growing emphasis on the in-situ characterization of chemical reactions, and efforts have been made to explore the dynamic changes in the material structures at different levels within the system under reaction conditions. These developments can be considered as the nascent stages of condensed matter chemistry research. Physics and chemistry have always been intertwined and mutually reinforcing natural sciences. Currently, new phenomena and theories in condensed matter physics continue to emerge. Introducing these new physical phenomena and theories into chemical research is a highly worthwhile exploration area. The present review will briefly introduce some relatively recent concepts in condensed matter physics (e.g., surface plasmon polariton, topological insulators, quasicrystals, local micro-electric/magnetic fields, light-matter interactions, alternating magnets, etc.) and their applications in chemistry. The aim is to illustrate the application potentials of cutting-edge condensed matter physics research in chemistry, provide insights into advancing traditional chemical research to the realm of condensed matter chemistry, and contribute to the development of condensed matter chemistry.
Contents
1 Introduction: from solid-state physics to condensed matter physics
2 Condensed matter in chemical reaction systems
2.1 Dynamic interface configurations in reactions
2.1.1 Reactions on solid-gas interfaces
2.1.2 Reactions on solid-liquid interfaces
2.1.3 Reactions on solid-solid interfaces
2.2 External electric field
2.3 Other external fields
2.4 Microenvironments
3 New chemical methods from the new concepts of condensed matter physics
3.1 Quantum confinement effects
3.2 Surface plasmon polariton
3.3 Topological insulator
4 Conclusion and outlook
Ran Jia, Chuipeng Kong, Wenfu Yan. Dimensions of Solid-State Material Systems and Their Chemical Reactions[J]. Progress in Chemistry, 2026, 38(1): 23-39.
With the continuous advancement and refinement of research methodologies, structural chemistry should evolve from the traditional exploration of “the relationship between reactants-products and chemical reactions” to the higher level of revealing and utilizing “the relationship between the dynamic structure of matter in condensed states and its chemical reactions”. When discussing chemical reactions within condensed matter systems, we cannot disregard the significant influences of the dynamic evolutions in their structures and the coupling of environmental factors. To elucidate the connection between the spatial dimensions of solid material systems and chemical reactions, we introduce the spatial dimension of crystal materials by starting from Bloch theory. Changes in spatial dimensions, surface configurations, and heterostructures can significantly alter their physical properties, thereby affecting the related chemical reactions, and even modifying the reaction pathways. In this review, we will discuss the ways in which spatial confinement effects can influence catalytic reactions, as demonstrated by the performance of carbon nanotubes in the asymmetric hydrogenation of ethyl propionate. Under reaction conditions, the intrinsic structure of solid surfaces, as well as the defects and catalyst particles distributed on them, undergo a series of dynamic changes. Beyond temperature and pressure, the environmental conditions of the reaction system (including pH-values, external electric/magnetic fields, optical fields, etc.) can also dynamically influence the geometric and electronic configurations of defects and catalytically active sites. Through a comprehensive introduction to these examples, we aim to clearly and concisely demonstrate how the dimensionality and dynamic changes in solid material structures affect chemical reactions, thereby emphasizing the importance and essentiality of research in condensed matter structural chemistry.
Contents
1 Introduction
2 Dimensions of solid-state systems
2.1 Reciprocal space and dimensions
2.2 Surface and edges
2.3 pH value
2.4 Low-dimensional materials
2.5 Reactions in confined spaces
3 Dimensions of defects and impurities
3.1 0D: Point defects
3.2 1D: Line defects
3.3 2D: Grain boundaries
4 Conclusion and outlook
Yihan Lu, Mengyu Xu, Yu Sun, Pu Zhao. Neutron Scattering Studies of Condensed Matter Chemistry[J]. Progress in Chemistry, 2026, 38(1): 40-58.
The advancement of characterization techniques has emerged as a pivotal driving force in refining structural theories of condensed matter chemistry. The unique interaction mechanism between neutrons and atomic nuclei/unpaired electrons enables neutron scattering techniques to provide distinctive information on light elements, isotopes, neighboring elements, and magnetism, thereby establishing complementary advantages with conventional optical, X-ray, and electron characterization approaches. Recent progress in high-flux neutron sources and in situ experimental methodologies has significantly expanded the application scope of neutron scattering from fundamental physics to complex chemical systems, making it an indispensable tool for deciphering intricate microstructures/microdynamics and reaction mechanisms. Among various techniques, neutron diffraction is mainly used to achieve a precise determination of both local and bulk structures. Concurrently, neutron spectroscopy offers unparalleled insights into dynamic processes and thus is particularly valuable for studying chemical bond breaking/formation, molecular conformation, molecule/ion diffusion/transport, and so on. Other neutron techniques, such as neutron imaging and small-angle neutron scattering, demonstrate huge potential for providing characteristic mesoscopic and macroscopic information. This comprehensive review systematically examines recent advances in neutron scattering investigations of condensed matter chemistry, with case studies underscoring the irreplaceability of these techniques in elucidating structure-property relations in complex systems. Furthermore, we present current challenges such as flux limitations, time resolution constraints, and multimodal characterization integration. We also propose forward-looking perspectives on methodological developments and synergistic characterization frameworks. These discussions offer valuable theoretical references and methodological guidance for researchers pursuing multi-scale characterization in condensed matter chemistry.
Contents
1 Introduction
2 A brief overview of neutron scattering techniques
3 Neutron diffraction studies of condensed matter chemistry
3.1 Single-crystal neutron diffraction studies
3.2 Neutron powder diffraction studies
3.3 Neutron pair distribution function studies
4 Neutron spectroscopy studies of condensed matter chemistry
4.1 Inelastic neutron scattering studies
4.2 Quasielastic neutron scattering studies
5 Neutron imaging/scattering studies of condensed matter chemistry
5.1 Neutron imaging studies
5.2 Small-angle neutron scattering studies
6 Challenges and opportunities
6.1 Technical challenges
6.2 Future research directions
7 Conclusion and outlook
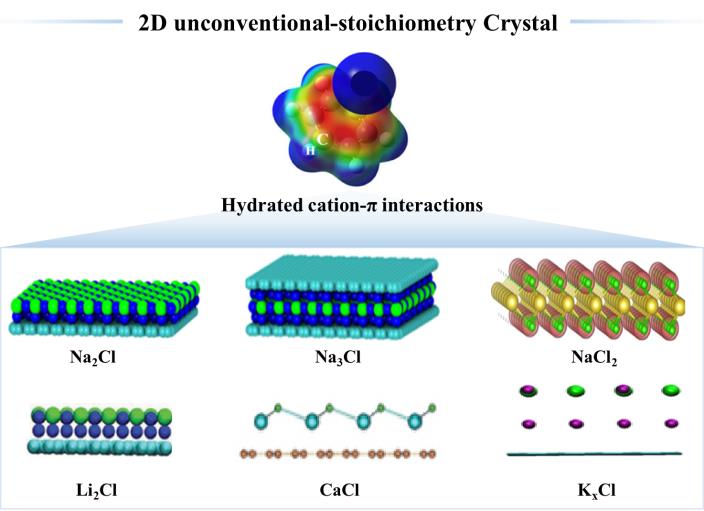
Mengjiao Wu, Xiaoling Lei, Haiping Fang. Two-Dimensional Crystal: From Na₂Cl to NaCl₂[J]. Progress in Chemistry, 2026, 38(1): 59-67.
The table salt we consume daily, sodium chloride crystal (NaCl), consists of one sodium atom for every chlorine atom. In fact, NaCl is the only crystal composed solely of sodium and chlorine elements that exists under normal temperature and pressure conditions. Recently, novel two-dimensional crystalline materials with unconventional stoichiometries, such as Na2Cl and Na3Cl, have successfully fabricated at ambient conditions. These two-dimensional (2D) crystals’ unique electronic structures endow them with novel attributes, which differ from those of conventional three-dimensional crystals. This review summarizes the recent progress made in the fabrication and analysis of the structures, distinctive features, and applications of these 2D unconventional-stoichiometry crystals Na2Cl、NaCl2、CaCl、K x Cl and Li2Cl on graphene surfaces in ambient conditions. Their special properties, including their piezoelectricity, metallicity, heterojunction, and room-temperature ferromagnetism, are paid particularly close attention. Finally, some significant prospects and further developments in this exciting interdisciplinary field are proposed.
Contents
1 Introduction
2 Hydrated cation- π interactions on the graphenebased material surface
3 Two-dimensional Na2Cl and Na3Cl crystals of unconventional stoichiometries in ambient conditions
4 Two-dimensional NaCl2 crystals of unconventional stoichiometries in ambient conditions
5 Two-dimensional CaCl, KxCl, Li2Cl, etc. crystals of unconventional stoichiometries in ambient conditions
6 Conclusion and outlook
Xiangyu Chen, Jianxin Kang, Lin Guo. Structure and Properties of Inorganic Amorphous Nanomaterials[J]. Progress in Chemistry, 2026, 38(1): 68-84.
Amorphous materials represent a vital component of condensed matter chemistry. Their atomic arrangement, which defined by long-range disorder and short-range order, confers distinct structural, physical, and chemical properties that differ significantly from those of conventional crystalline materials. This paper, focusing on inorganic amorphous nanomaterials (ANMs), first examines the characteristics of amorphous structures from a microscopic perspective, with an emphasis on the types and mechanisms of chemical bonds as well as intermolecular interactions within these materials. Subsequently, from a macroscopic application-oriented perspective, it discusses in detail the pivotal roles and underlying mechanisms of amorphous structures in regulating material properties, chemical reaction processes, and functional applications. Then, based on a multi-scale structural perspective, the work conducts an in-depth analysis of the critical contributions of their electronic structures, defect characteristics, and amorphous disordered structure design to chemical reactions, while aiming to establish the structure-activity relationship between amorphous structures and reaction activity. Finally, it outlines the research directions and application prospects of amorphous materials and their structural features, providing a systematic reference framework for advanced studies in the field of condensed matter chemistry.
Contents
1 Introduction
2 Concepts of amorphous materials
3 Amorphous state structural chemistry
3.1 Interaction forces of amorphous materials
3.2 Role and influence of amorphous structure
3.3 Multiscale structure of amorphous state and chemical reactions
4 Conclusion and outlook
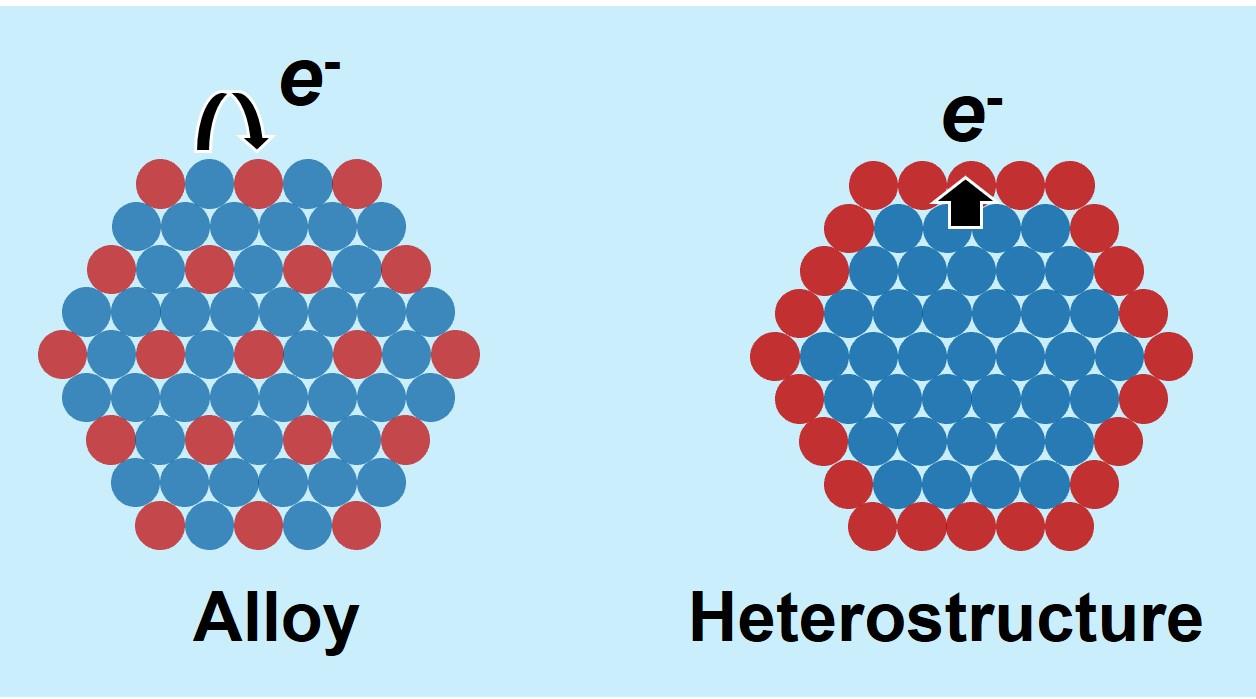
Zhaojun Liu, Chuanbo Gao. Control of Atomic Condensed States in Multimetallic Nanoparticles and Their Catalytic Effects[J]. Progress in Chemistry, 2026, 38(1): 85-102.
In nanoscale metal particles, atoms of different elements can exhibit various condensed states: they may either fully mix to form homogeneous alloys or separate into distinct phases, creating heterogeneous structures. These diverse atomic arrangements significantly affect the electronic coupling and catalytic properties of the multimetallic nanoparticles. Precise control over the atomic condensed states within nanoparticles holds the promise of optimizing their electronic structures, offering significant opportunities for the design of novel nanocatalysts with distinctive properties. However, controlling atomic condensed states in nanoparticles using current wet-chemical methods remains challenging. In the synthesis of alloy nanomaterials, the intrinsic reduction potential differences between metal salts cause significant variations in reduction kinetics, making it difficult to achieve uniform alloying and precise control over the alloy compositions. In the synthesis of heterogeneous structures, the reduction potential differences cause galvanic replacement reactions between noble metal salts and less-stable metal nanostructures, limiting the controllability of nanocrystal growth. This paper reviews recent research progress in overcoming these synthesis limitations for controlled atomic condensed states of metal atoms in nanoparticles. Specifically, introducing an active hydrogen (i.e., hydrogen atoms or radicals) interfacial reduction mechanism has mitigated the impact of reduction potential differences, improving the mixing homogeneity of different metal atoms within nanoparticles. This approach also allows precise control over the content of each metal component within nanoparticles. By modulating the reduction potentials of metal salts, it has become possible to suppress the galvanic replacement reaction between noble metal salts and less-stable metal nanostructures, leading to a novel family of core-shell nanostructures with a less-stable metal core and a noble metal shell. By precisely regulating the atomic condensed states within multimetallic metal nanoparticles, researchers have been able to effectively tune the electronic structures of these materials, significantly improving the catalytic performance. These advancements highlight the potential of controlled atomic condensed states in multimetallic nanoparticles for developing high-performance catalysts across various applications.
Contents
1 Introduction
2 Homogeneous alloy nanomaterials
2.1 Conventional wet-chemical synthesis: challenges in controlling atomic condensed states
2.2 H·-involved interfacial reduction strategy
2.3 Vacancy-diffusion-induced alloying approach
2.4 Nanoreactor-based synthesis of alloy nanoclusters
3 Heterostructured multimetallic nanomaterials
3.1 Conventional core-shell nanostructure synthesis:limitations of metal activity series on core-shell structures
3.2 Synthesis of core-shell nanostructures with inverse metal activity series
4 Catalytic regulation based on atomic condensed states in multimetallic nanoparticles
4.1 Electronic coupling effect
4.2 Strain effect
4.3 Geometric effect
5 Conclusion and outlook
Yufei Wang, Xiang Wang, Dong Wang. Electrochemical Interfacial Structures and Processes: Perspective from Condensed Matter Chemistry[J]. Progress in Chemistry, 2026, 38(1): 103-115.
Electrochemical systems are composed of fundamental components such as electrodes and electrolytes, whose composition, phase, and structure have an important influence on the electrochemical performances. The electrochemical interface serves as the core zone for species transformation, charge transfer and electrochemical reactions. With the development of advanced in-situ electrochemical characterization techniques, in-depth investigation and understanding of the dynamic processes at the electrochemical interface are essential for the precise construction of high-performance systems. In this review, we present a systematic summary of electrochemical interfacial processes and characterization from the perspective of condensed matter chemistry. The basic components of electrochemical systems, such as electrodes and electrolytes, are introduced, and the characteristics of electrochemical interfaces in view of condensed matter chemistry are discussed. The characterization methods and techniques for electrochemical interfaces are summarized. In addition, the regulations of some electrochemical dynamic processes are re-examined.
Content
1 Introduction
2 Condensed matter systems in electrochemistry
2.1 Electrode
2.2 Electrolyte
2.3 Dispersed medium
3 Understanding electrochemical interface from a condensed matter chemistry perspective
3.1 Integrality
3.2 Dynamics
3.3 Multi-scale
4 Electrochemical interfacial characterization techniques
4.1 Imaging characterizations of interfacial structures of condensed matter
4.2 Spectroscopic characterizations of species in condensed matter systems
5 Electrochemical interfacial processes and properties
5.1 Interfacial structure evolution and characterization
5.2 Interfacial process regulation
6 Conclusion and outlook
Zhiyuan Xing, Wenjun Yang, Haoqi Shi, Yang Peng. Metal-Organic Diffusion Layer (MODL) in Electrocatalytic Carbon Dioxide Reduction Chemistry[J]. Progress in Chemistry, 2026, 38(1): 116-129.
Electrochemical CO2 reduction (eCO2R), as one of the pivotal technologies for achieving carbon neutrality strategic goals, demonstrates significant application potential in renewable energy storage and high-value chemical synthesis. The electrode-electrolyte interfacial electric double layer (EDL), serving as the highly active reaction zone, profoundly governs the overall system performance by coupling reaction kinetics at catalytic sites with interfacial mass transport processes. Conventional solid/liquid EDL systems suffer from limitations imposed by one-dimensional regulatory mechanisms relying on electric field-driven effects and static interfacial configurations, resulting in restricted ion spatial distributions and insufficient dynamic regulation dimensions. This inherent constraint hinders the synergistic optimization of interfacial reaction kinetics and mass transport processes. To address these challenges, we propose the construction of a "Metal-Organic Diffusion Layer" (MODL) architecture. Through molecular design strategies incorporating functionalized organic components (e.g., amphiphilic molecules, coordinating and charged polymers), this approach leverages their abundant functional groups and dynamic interfacial characteristics to precisely regulate the hierarchical condensed-phase structures within the MODL at the near-field microscopic scale (e.g., electrode crystallinity, reaction pathways), far-field mesoscopic scale (e.g., interfacial electric field, water molecular configuration), and macroscopic scale (e.g., interfacial wettability, mass transport channels), achieving precise regulation of spatial partitioning of electrode interfaces. This work will systematically analyze the organic-mediated dynamic coupling mechanism between the near-field catalytic core and far-field mass transport environment, elucidating their synergistic interplay in regulating CO2 conversion pathways and interfacial kinetics. The established multi-dimensional MODL interfacial model provides a theoretical framework for deciphering intricate structure-performance relationships in electrochemical interfaces, laying scientific foundations for the rational design of efficient and stable eCO2R catalytic systems.
Contents
1 Introduction
2 Linking metal-organic diffusion layer configurations to eCO2R performance
2.1 Near-field interfacial engineering: organic component-induced reactive crystalline surface exposure and reaction path modulation
2.2 Far-field mesoscale regulation: organic-mediated structural expansion of diffusion layer and gradient microenvironment reconstruction
2.2.1 Far-field regulated water/electric field structures,local pH
2.2.2 Far-field modulation of substance transport
3 Conclusion and outlook

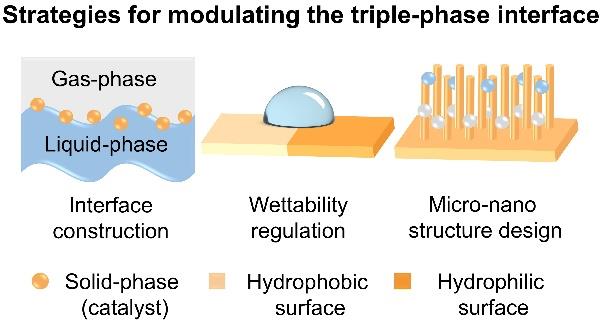
Lanze Li, Jiexin Wen, Shengbo Zhang, Chenxi Ma, Jun Yin, Qiong Lei. Triple-Phase Interface Engineering in Electrocatalytic CO2 Reduction[J]. Progress in Chemistry, 2026, 38(1): 130-140.
Catalytic materials offer multidimensional tunability for catalytic reactions, owing to their multi-level structural features in the condensed state, such as defect types, surface and interface configurations, and wettability. Aimed at maximizing reactive interfaces and enhancing catalytic efficiency, modulating hierarchical phase structures has emerged as a pivotal strategy for performance optimization in catalytic systems. The electrocatalytic CO2 reduction reaction (eCO2RR) system, as a typical solid-liquid-gas triple-phase interface reaction, inherently exhibits reaction kinetics and catalytic performance governed by the spatial distribution and dynamic characteristics of the triple-phase interface on catalyst surfaces. Therefore, the regulation of the triple-phase interface to maximize the reaction interface is a potent pathway to enhance the catalytic performance of eCO2RR. This perspective systematically reviews the evolution from traditional liquid-solid biphasic interfaces to advanced solid-liquid-gas triple-phase interfaces delves into the primary challenges of constructing stable triple-phase interfaces on gas diffusion electrodes within the eCO2RR system and summarizes the latest advancements in regulating the triple-phase interface by enhancing hydrophobicity. Achieving a fine balance between hydrophobicity and hydrophilicity (i.e., optimal wettability) is crucial for constructing an efficient triple-phase interface. On one hand, sufficient hydrophobicity is required to prevent excessive electrolyte infiltration: on the other hand, moderate hydrophilicity must be maintained to ensure the supply of reactants/ions from the electrolyte. Their dynamic equilibrium can significantly optimize the triple-phase interface structure, enhance mass transfer of reactants, and improve the effective utilization of catalytic active sites. These insights provide valuable guidelines for designing high-performance triple-phase interfaces in eCO2RR and other gas-involving electrochemical processes, thereby promoting the development of sustainable energy technologies.
Contents
1 Introduction
2 Condensed matter chemistry and eCO2RR
3 Evolution of solid-liquid-gas interfaces and their challenges in catalysis
4 Condensed-matter-based strategies for triple-phase interface regulation in eCO2RR
4.1 Triple-phase interface construction
4.2 Wettability regulation
4.3 Micro⁃nano structure design
5 Conclusion and outlook
Shaokang Shi, Li Zhao, Zhongyuan Lü. Condensed Matter Chemistry in Macromolecular Phase Separation and Self-Assembly[J]. Progress in Chemistry, 2026, 38(1): 141-150.
This review summarizes recent advances in condensed matter chemistry within the field of macromolecular phase separation and self-assembly, with emphasis on polymer structure and morphology regulation, functional mechanisms of biomolecular condensates, and the application of multiscale theoretical simulations. Studies have shown that the hierarchical structures of polymers are highly sensitive to reaction conditions. Polymerization-induced phase separation and self-assembly represent key strategies for controlling structural evolution, while the coupling of kinetic factors plays a pivotal role in pattern formation. In biological systems, liquid–liquid phase separation of intrinsically disordered proteins and the subsequent formation of condensates are critical for cellular function. These condensates not only regulate biochemical reactions within their compartments but also undergo feedback regulation by these reactions, thereby forming complex dynamic networks. These intricately coupled processes call for more advanced methodologies. In this context, multiscale theoretical simulations, particularly hybrid approaches that integrate molecular dynamics and Monte Carlo methods, provide powerful tools to probe the formation and evolution of hierarchical structures under varying reaction conditions. Despite significant progress, several challenges remain. For instance, precise control over the driving forces of polymer phase separation and high-fidelity simulations of biomolecular condensate structure and function continue to be pressing issues. Future studies should focus on elucidating the dynamic effects of condensed-phase reactions on polymer hierarchical structures, systematically dissecting the structural regulation of biomolecular condensates, and further exploring the impact of chemical modifications on their multiscale organization. Such endeavors will not only deepen our understanding of the fundamental principles of condensed matter chemistry but also provide new theoretical support and research directions for the advancement of this field.
Contents
1 Introduction
2 Condensed phase structures and morphologies of polymers
2.1 Structures and morphologies of polymers
2.2 Mechanisms of polymerization-induced phase separation( PIPS) and self-assembly( PISA)
2.3 Role of kinetic coupling in pattern formation
3 Biomolecular condensates and functional phase separation
3.1 Liquid-liquid phase separation (LLPS) of intrinsically disordered proteins
3.2 Formation and function of biomolecular condensates
3.3 Feedback between biochemical reactions and condensate formation
4 Multiscale theoretical modeling approaches
4.1 Coarse-grained modeling of macromolecular systems
4.2 Hybrid molecular dynamics/Monte Carlo (MD/MC)techniques
4.3 Simulation of structure evolution under condensedphase conditions
4.4 Modeling challenges in reactive and biological systems
5 Challenges and future perspectives
Bao Li, Mengjie Liu, Lixin Wu. Condensed Matter Chemistry in Gel Systems: From Preparation and Structure to Physicochemical Properties and Material Applications[J]. Progress in Chemistry, 2026, 38(1): 151-162.
Condensed matter chemistry possesses rich connotations and extensive potential for expansion, offering novel perspectives and insights into the understanding and recognition across multiple domains of chemistry. While its application in solid and liquid systems has been elucidated to some extent, further exploration and strengthening are required in broader chemical research fields and material states. As a substance intermediate between liquid and solid phases, gels exhibit multi-level network structures, diverse physical and chemical properties, and significant application prospects, making them an ideal candidate system for condensed matter chemistry investigations. From the perspective of condensed matter chemistry, this paper systematically expounds on the principles and applications of condensed matter chemistry within gel systems, as well as their mutual validation relationship, by exploring fundamental concepts and research contents in gel systems. Specific topics include: the application of condensed matter chemistry in gel preparation strategies and the resulting structural transformations: the multi-level structure of gels, ranging from microscopic atomic and molecular arrangements to mesoscopic nanoscale structures and macroscopic material configurations, along with their interrelationships: characterization methods and technologies in gel research and their correlation with gel structures: the utilization of condensed matter chemistry to interpret the physical and chemical properties of gels and the pathways and mechanisms of chemical reactions within gel systems: the relationship between gel material structure and performance, as well as interactions among components in complex systems: and typical applications of gel materials in tissue engineering, drug delivery, human-machine interfaces, and environmental science. The systematic elaboration and summary of these contents will enhance the understanding of condensed matter chemistry’s role in gel systems and provide theoretical foundations for the design and optimization of high-performance gel materials.
Contents
1 Introduction
2 Preparation strategies of gels and condensed matter chemistry
3 The multi-level networks of gels and the characterization methods and techniques
3.1 The structures of gels and condensed matter chemistry
3.2 Characterization methods and techniques of gel structures
4 The physicochemical properties of gels and chemical reactions in gels
4.1 The physicochemical properties of gels
4.2 Chemical reactions in gels
5 The applications of gels
5.1 The applications of gels in tissue engineering
5.2 The applications of gels in drug delivery
5.3 The applications of gels in human-machine interface
5.4 The applications of gels in water treatment
6 Conclusion and outlook
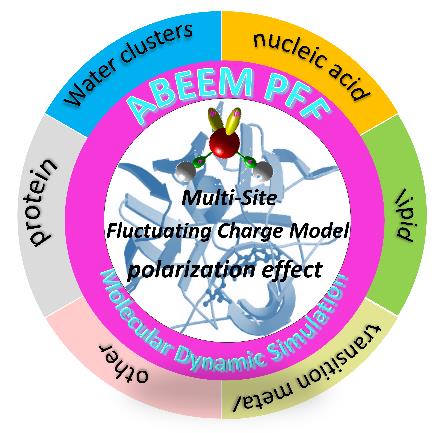
Dongxia Zhao, Qingyan Sun, Cui Liu, Jian Zhao, Lidong Gong, Zhongzhi Yang. Molecular Dynamics Simulation of Biomolecular Aqueous Systems[J]. Progress in Chemistry, 2026, 38(1): 163-180.
Biomolecular aqueous solutions represent a crucial class of condensed matter and serve as the fundamental basis of living organisms. Investigating their structural and chemical properties holds immense scientific and practical significance. Molecular dynamics (MD) simulations are a powerful tool for studying biomolecular systems, where accuracy critically depends on the precision of the molecular force field. Traditional force fields employ fixed atomic charges, neglecting polarization effects and charge transfer. Over recent decades, significant efforts have been devoted to developing polarizable force fields. The atom-bond electronegativity equalization method (ABEEM) polarizable force field effectively captures molecular polarization and charge transfer. This review outlines the ABEEM methodology, with emphasis on the ABEEM-7P water model and the application of the ABEEM polarizable force field is provided. The work has carried out using both the ABEEM-7P water model and the ABEEM polarizable molecular force field for molecular dynamics simulations of biomolecule aqueous systems,and the results have been given by representative case studies.
Contents
1 Introduction
2 Theoretical methods overview
2.1 Born-oppenheimer approximation for solving the time-independent Schrödinger equation
2.2 Molecular mechanics (MM)
2.3 Common force fields
2.4 ABEEM polarizable force field
2.5 Molecular dynamic simulation
3 Molecular dynamics simulation in biomolecular aqueous solution system
3.1 Application of ABEEM-7P water model and polarization force field
3.2 Molecular dynamics simulation example of ABEEM PF biomolecule aqueous solution system
4 Conclusion and outlook