




1 引言
锂作为最轻的碱金属,具有许多优异的理化性质,如高的电化学活性、高比热容和高的氧化还原电势等。这些特性使得锂及其化合物一直备受人们关注。近年来,锂已经广泛应用于电池、电子信息设备、陶瓷、航空航天、冶金工程等诸多高科技领域,被认为是21世纪民生经济和国防建设的重要战略资源[1]。
随着新能源产业的蓬勃发展,全球锂资源消费从2016年的22.8万吨LCE(碳酸锂当量)迅速增长到2022年的78.0万吨LCE[2],2023年的消费量预计将达到约107万吨LCE[3]。从长远角度来看,未来锂资源的需求量还将持续快速增长,但由于受到锂矿开采条件、提锂技术等限制,全球锂资源市场高需求、低供给的现状不可避免。据调查[4,5],大陆卤水是锂赋存的最大资源(64%),其次是硬岩(36%)。因此,从水资源中回收锂极为必要,并已成为行业发展新趋势。值得注意的是,中国是世界最大锂消费国,锂资源占全球6.31%,居世界第五[6],但其三分之二蕴藏在低品位的盐湖卤水中,普遍存在镁锂比高、大量碱金属和碱土金属共存等现象,给提锂带来了困难[7]。且锂资源供需关系极度失衡,导致电池级碳酸锂(Li2CO3)价格从2022年底冲高至59.5万元/吨而2023年回落到22.4万元/吨[8],目前又显现出止跌又回升的趋势,影响行业健康发展。因此,寻求环保、低成本和高选择性提取锂技术并建立锂资源回收体系具有重要的理论与实际意义。
目前,从卤水中回收锂的常见方法包括溶剂萃取[9]、沉淀结晶[10]、吸附[11,12]、膜分离[13]等。其中,沉淀结晶法在提取锂方面应用较早,其成熟的沉淀技术适合低镁锂比体系。但该方法选择性较差,不适合我国高镁锂比的盐湖体系。溶剂萃取法由于其在湿法冶金方面有着成熟的应用基础,在高镁锂比盐湖体系中提取锂应用较多,至今仍然是选择性分离和金属提取的主要方法之一。但溶剂萃取易产生三相,使用的有机溶剂会腐蚀工艺设备,且溶液泄漏存在污染环境的隐患,严重阻碍了该方法的推广应用。膜分离技术包括选择电渗析(SED)[14]、纳滤(NF)[15]、离子印迹膜(IIM)[16]和膜电容去离子(MCDI)[17]等,具有高效、环保等优点,在锂回收领域显示出巨大的应用潜力,但其能耗高、膜耐久性不理想等缺点限制了它们的工业化。
相比于上述提锂技术,吸附法凭借其成本低、操作简单、绿色环保、高选择性和适用范围宽等突出优势,近年来得到迅速发展,尤其适用于我国大体积、低浓度目标离子的液相体系。吸附法是通过选择或设计合成对Li+有识别能力的吸附材料,利用特定的物理或化学作用,实现对锂离子选择性分离回收效果。吸附材料是吸附提锂的核心。一般而言,理想的吸附材料应具备以下特点:1)高的选择性,能够从元素组成复杂且盐度高的溶液中选择性捕获Li+; 2)良好的稳定性,在高盐度和反复吸附/解吸过程中保持良好的结构稳定性和机械稳定性;3)足够的吸附容量,即单位质量的吸附材料能够尽可能多捕获锂离子。目前,吸附材料大体可分为有机、无机以及复合吸附材料(图1 )。其中,有机吸附材料包括冠醚、杯芳烃类等,前者的选择性主要是依靠调控空穴尺寸与目标离子适配,以及环上杂原子种类等因素。后者与Li+的配位性和溶剂密切相关,由于其在水溶剂中溶解度有限,目前对碱金属选择性的研究主要围绕甲醇、四氢呋喃等溶液[18]。无机吸附材料主要分为铝基吸附剂、锂离子筛、天然矿石和碳材料。天然矿石以及碳材料的表面活性位点可以与大多数金属结合,对Li+选择性较差且吸附容量低,限制其在卤水提锂的应用。锂离子筛类吸附剂具有良好的吸附效率、吸附选择性和化学稳定性,主要包括锂锰氧化物(LMO)以及锂钛氧化物(LTO)。锂锰氧化物由于酸浸过程中溶损高、循环稳定性能差、平衡速率慢等问题限制发展,可以通过金属元素掺杂[19]、结构形态的改变[20]、改善制备方法[21]来提高其性能。与锂锰氧化物相比,锂钛氧化物金属溶损率低、稳定性能好,应用前景较好。铝基吸附剂已取得商业化运用,具有成本低、吸附性能稳定等优点。以铝基吸附剂为例,吸附材料整体提锂工艺大致如下:Li+被吸附饱和后,对吸附剂进行水浸,使锂、铝分离。再使用沉淀剂去除含锂溶液中的镁、钙等杂质,蒸发浓缩后加入碳酸钠进行沉锂反应,实现碳酸锂产品的生产。本文主要围绕不同类型吸附剂研究进展进行综述,横向比较不同吸附剂的制备方法、吸附性能与吸附机制,为后续研究新型提锂吸附剂、优化制备技术和推动提锂技术在盐湖卤水中的应用提供有益参考。
2 冠醚类吸附剂
2.1 冠醚吸附剂的制备
冠醚(Crown ether, CE)又称为大环醚,是由多个氧原子和碳原子组成的环状有机化合物。由于其“大环效应”,具有富电子空腔的CE可以与特定的金属离子形成非常稳定的络合物[22]。冠醚对阳离子具有精确的尺寸选择性,精确性取决于空腔尺寸和金属离子直径的接近程度。通常,空腔尺寸和目标离子直径越接近,目标离子被选择性捕获概率越高。大量研究表明,具有12~14元环的冠醚对Li+有具有很强的结合亲和力[23]。然而,冠醚价格昂贵且回收困难,往往通过接枝或聚合到纤维、有机树脂、多孔大分子材料等基体上形成有机配体复合类吸附剂。这种复合吸附剂不仅可以保留配体对Li+特异性识别能力,而且兼具基体的高稳定性、耐酸性、易于固液分离等性质,进而能够显著提高CE的可接触性和循环使用等性能。涉及的基体材料有硅类材料[24]、磁性颗粒[25]、多孔材料[26]等。基体材料作为吸附剂的骨架,连接冠醚配体实现Li+配位。从实际应用的角度,基底材料必须具备机械强度高、耐酸洗、稳定性好、易于嫁接修饰的优点。
Bai等[27]通过高内相乳液(HIPEs)模板法制备了多孔聚合物(PolyHIPEs),利用紫外线(UV)引发表面聚合从而引入2-(烯丙氧基)甲基-12-冠-4(2AM12C4),合成官能化聚合物刷(PVBC-g-PCE)。PVBC-g-PCE具有物理强度高、可接触结合位点密度高、孔隙高度可渗透等优点,是一种用于选择性捕获Li+的良好吸附剂。Tachibana等[24]制备了嵌入高孔二氧化硅珠中的冠醚型有机复合吸附剂,如苯并-15-冠-5(BC15)、苯并-18-冠-6(BC18)等,对海水中的锂离子具有相对较高的吸附能力。Cheng等[26]利用绿色的低温相分离方法制备壳聚糖(CS)纳米纤维膜,选择具有独特空腔结构的2-羟甲基-12-冠-4(2H12C4)接枝到CS表面,制备出冠醚修饰的壳聚糖纳米纤维膜(CS-CE)。该类材料吸附容量大、循环稳定性好,且具有良好的选择性,在锂离子提取方面表现出良好的应用潜力。Jo等[28]将作为离子受体的大环螯合配体(12C4)分子并入聚醚砜(PES)纳米纤维膜的石墨烯中(CGPNF),CGPNF膜显示出86.3 mg/g的最大吸附容量并且在10次再生循环后保持其初始吸附容量的93%以上。Ding等[29]通过化学接枝和静电纺丝将2-甲基-12冠-4(2M12C4)接枝到纳米SiO2上,合成纳米级吸附材料PAN-CE@SiO2。该材料具有低成本、良好的流体力学性能,并且经证明可以选择性地吸附Li(I)。
2.2 选择性提锂性能
冠醚吸附剂在提取海水、盐湖卤水等水资源的Li+应用中,最受研究者关注的是其选择性、吸附容量以及循环稳定性等性能。
1) 冠醚单体不同,对锂离子识别效果不同。Dixit等[30]通过三种不同肌醇取代基键合12-冠-4,发现不同肌醇取代基对Li+的识别性能有明显差异,可以通过改变冠醚氧原子的相对取向来调节金属离子与冠醚的相对结合亲和力。Tachibana等[24]实现了高孔硅珠包埋的冠醚型有机复合吸附剂的合成,用于同时回收海水中的锂和铀。合成的冠醚树脂包括苯并-12-冠-4(BC12)、二苯并-14-冠-4(DBC14)、苯并-15-冠-5(BC15)、苯并-18-冠-6(BC18)等,发现不同冠醚单体,吸附性能有区别。BC15和BC18树脂对海水中Li+具有较高的选择性吸附能力,其吸附行为强烈依赖于这些冠醚空腔大小和疏水程度。Park等[31]使用甲基冠醚(AC-SBA-15)和氮杂冠醚(HMC-SBA-15)部分官能化介孔二氧化硅材料,不同冠醚基团官能化的材料均能在人工海水中实现高选择性地吸附Li+。
2) 基体材料的类型、比表面积、亲水性、吸附位点数目等因素对冠醚选择性吸附锂离子的行为具有重要影响。Zheng等[32]用冠醚官能化氧化石墨烯(GO)、壳聚糖和聚乙烯醇(PVA),成功制备了系列纳米纤维膜,最大吸附量可达168.50 mg/g。在选择性吸附实验中,纳米纤维膜对盐湖卤水中的Li+具有高选择性。5次循环后,材料仍保持88.31%的吸附容量。虽然有机复合吸附材料具有明显选择性和稳定性,但是活性材料较高的质量与锂较低的相对原子质量存在矛盾并且有机聚合物交联骨架包埋内部活性位点等问题,限制了吸附容量的发展。此外,还有SiO2、Fe3O4、碳纳米管、玻璃纤维垫都是适用于负载CE的良好基体材料[33]。研究表明,具有高比表面积的基材,能够加快传质速率,进而增强吸附剂的吸附性能[34]。Wang等[35]利用Pickering乳液模板法,在乳液中引入冠醚单体合成冠醚功能化的多孔基体材料,用于锂离子选择分离。其开发的基体材料的多孔性质以及大量的活性位点,有利于提升吸附材料的吸附容量。Alexandratos等[36]通过化学接枝法在聚甲基丙烯酸缩水甘油酯基体表面上固定14-冠-4,定量分析了水溶液中Li(I)的选择性。并且实验结果表明冠醚络合Li(I)的程度受聚合物基质极性的影响,随着基质亲水性的增加,冠醚对Li(I)的络合能力越强。上述部分冠醚吸附材料的性能总结如表1 。
表1 冠醚配体复合吸附材料的性能Table 1 Performance of crown ether ligand composite adsorption materials |
| Crown ether ligands | Matrixes | Specific surface area (m2/g) | pH | Adsorption capacity (mg/g) | Selectivity α(Li/Na) | Cycling stability | ref |
|---|---|---|---|---|---|---|---|
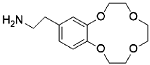 Aminoethylbenzo-12-crown-4 | Polymer nanosheets (PMBA-PMA) | / | 7 | 14.67 | / | 90% (Five cycles) | 30 |
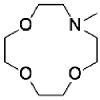 1-aza-12-crown-4 | mesoporous silica SBA-15 3-Aminopropyltriethoxy 4-ylsilane | 578 | 8 | 7.63 × 10-3 | / | / | 31 |
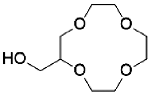 2- (hydroxymethyl) 12-crown-4 | graphene oxide chitosan polyvinyl alcohol | 101.5 | 7 | 168.50 | 2.51 | 88.31% (Five cycles) | 32 |
 Aminoethylbenzo-12-crown-4 | Porous polymer substrate (PVBC) | / | 7 | 4.22 | 6.59 | 95.0% (Five cycles) | 35 |
 Octamethyl 14-crown-4 | methacrylate polymer | / | / | 3.05 | / | / | 36 |
1) 选择性系数α,也称分离因子,是表示某一单元分离操作或某一分离流程将两种物质分离的程度,其计算方法为 =Kd(Li+)/Kd(Me)。Me: K+、Na+、Ca2+、Mg2+。 2) Kd为吸附分配系数,是指一定温度达到反应平衡时,组分在固定相中的质量分数与流动相中的质量浓度之比,其大小反映离子在固液两相中的移动与分离能力。 |
2.3 选择性提锂机理
2.3.1 冠醚空穴尺寸与离子尺寸的匹配程度
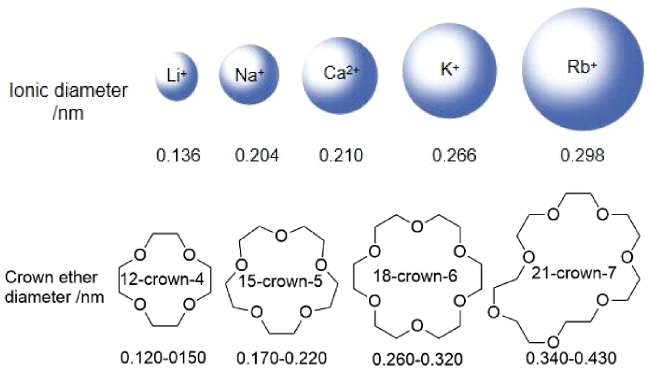
冠醚型吸附剂的选择性受到冠醚孔空穴尺寸与离子尺寸的匹配程度的影响。由于醚键中氧原子提供电子的作用,主体可以与客体离子形成配位。空腔尺寸与目标离子直径越接近,其进入冠醚空穴的可能性越大。小环冠醚(C*-C*)空腔尺寸通常在0.122~0.153nm之间,而锂离子尺寸为0.118nm,Li+完美贴合空腔尺寸从而形成捕获,并且可以与空腔中的醚键形成强烈的静电相互吸引。而K+、Na+、Ca2+等离子直径远大于Li+,它们难以与小环冠醚稳定结合。图2 中列出部分碱金属离子直径以及冠醚尺寸[37]。
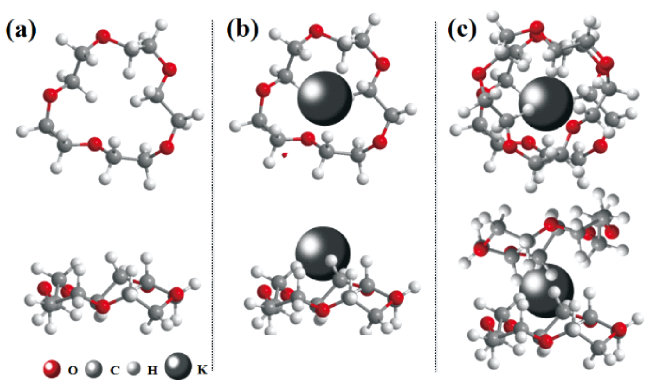
Valente等[38]发现随着冠醚上氧原子数目从4增加至12,Li+的分离系数变化不大,而对其他碱金属K+、Rb+、Cs+分离系数显著增加。含4个氧原子的冠醚就可以使得Li+的第一配位层得到饱和,而K+、Rb+、Cs+所需电子更多,需要更多氧提供电子。所以选择小环的冠醚,将有利于Li+选择性分离。值得一提的是,直径较大的金属阳离子会在氧原子构成的空穴平面上方配位,形成金字塔结构。另一种情况,阳离子位于空腔上方,阳离子将两个冠醚进行桥联,形成夹层结构,如图3 所示。夹层结构相对金字塔结构,其阳离子配位不紧密、稳定性较差[39]。当金属离子较小时,配体发生畸变,将阳离子包围在其中,形成包合物以减少溶剂进入,但是离子距离配体原子远、静电引力小。所以当金属离子与空腔尺寸刚好适合的冠醚形成立体匹配时,吸附材料才具有突出特异选择性。
2.3.2 冠醚分子的柔软程度
冠醚分子的柔软程度对选择性也有影响。当冠醚分子刚性比较大时,冠醚专一性高,仅对单一的离子具有选择性。但事实上,目前文献报道的大多数冠醚分子比较柔软,其空穴尺寸容易发生变化,从而适应一定尺寸范围的多种金属阳离子[40]。Taziaux等[41]报道了一系列基于单氮杂冠醚连接的香豆素343的荧光离子载体的光物理和络合性质,在先前实验中,研究了由香豆素C343通过酰胺桥连接1-氮杂-15-冠-5(C343-crown)组成的荧光离子载体的光物理性质和络合性质,发现C343-crown对碱土金属阳离子的选择性更好。为了改变荧光探针的选择性,使用单氮杂-15-苯并冠-5和单氮杂-15-二苯并冠-5取代了单氮杂-15-冠-5。结果表明,通过引入苯并基团对络合腔的刚性化,大大提高了碱土金属离子对Li+检测的选择性。Babujohn等[42]报道了一种新型的基于三苯乙烯的芳香连接,通过柔性苯冠单元的氧化三聚化,一步形成高结晶的COFs/COP。采用了商业化的二苯并-18-冠-6和合成的二苯并-24-冠-8的氧化偶联,得到的相应共价有机框架(COFs)/共价有机聚合(COP)分别记为COP-TPC6和COP-TPC8。冠醚单体的柔韧性有望提供额外的优势,如客体结合时的可逆结构动力学。考虑到冠醚通过O-Au相互作用对金离子具有很强的亲和力,探索了COP-TPC8和COP-TPC6作为吸附剂从溶液中回收Au3+离子。吸附研究表明,COP-TPC8和COP-TPC6具有较好的吸附容量及Au3+高选择性。
2.3.3 供体杂原子以及取代基
除了空穴尺寸以及冠醚分子柔软程度影响之外,冠醚环中起配位作用的杂原子类型和数量、环上取代基的种类以及离子的电荷与尺寸比亦会影响主体冠醚和客体金属阳离子之间的相互作用。梁苏卓成等[43]基于密度泛函理论研究了Si掺杂15-冠-5,发现Si可以增大冠醚尺寸,并且通过不同掺杂方式增强/减弱Li+识别能力。依据自然布居分析显示,Si掺杂冠醚尽量避免环中出现Si—O—Si键,该单元使得O难以极化Si的电子,并且带正电的Si和Li+距离很近,出现静电排斥现象不利于冠醚络合Li+。Torrejos等[44]以羧基(—COOH)作为冠醚环氧封端接头的官能化位点,发现可以提升Li+的选择性吸附容量。基于此,他们制备由羧基作为侧臂的冠醚固体负载吸附剂。其中,类型1为中性羟基-二苯并-14-冠-4醚(HDB14C4)固定的多壁碳纳米管(MWCNTs),类型2为带有HDB14C4—COOH侧臂的MWCNTs。他们发现类型2中—COOH侧臂的存在显著增强了pH=7下对Li+的吸附容量,且在竞争环境中呈现出Li+>Na+>Mg2+>Ca2+>K+>Sr2+的选择性。
3 铝基吸附剂
3.1 铝基吸附剂的制备
铝基吸附剂属于金属基吸附剂中特殊的一类吸附剂。该吸附剂基本骨架主要由氢氧化铝组成,所以称为铝基吸附剂,是目前唯一投入工业化应用的吸附材料。其中的Li-Al层状双氢氧化物(LiAl-LDHs)是目前广泛研究的铝基吸附剂,综合性能突出,包括可忽略的洗脱损伤、制备工艺简单、绿色经济、稳定的吸附性能等。
目前常见的吸附表征手段主要为静态吸/脱附,静态吸/脱附是指溶液在不流动条件下,定量吸附剂和定量溶液经过长时间充分接触而达到吸附平衡,得到平衡吸附容量;动态吸/脱附是指某一浓度的溶液以一定流速和温度下流过填充有一定质量吸附剂的吸附柱,从而获得透过吸附容量和平衡吸附容量。
郭敏等[49]分别通过一步法和浸泡法制备铝基吸附剂,进行Li+静态吸/脱附和动态吸/脱附性能研究,发现在这两种情况下该吸附剂均有较好的吸附性能。选择性次序为Li+>Mg2+>Na+>K+,吸/脱附前后结构稳定,基体本身未发生溶损。Liu等[50]通过机械球磨制备了LiCl·2Al(OH)3·xH2O铝基材料,该材料对锂的沉淀率达78.3%,且能够有效分离锂和镁,沉淀物中Mg/Li质量比仅为0.02。Zhong等[51]通过简单的共沉淀方法制备具有规则二维六边形平面的Li/Al LDH,对Li+具有很强的选择吸附能力,经过12次吸附-解吸循环,Li/Al LDHs的吸附容量仍超过7.0 mg/g。董茜等[52]通过LiOH浸泡Al(OH)3后进行酸转化,制备出含特定空穴的Al(OH)3结晶。该吸附剂锂吸附量达到0.6~0.9 mg/g,在卤水中对Li+选择性高,Mg2+、K+等碱金属元素基本不吸附。
3.2 铝基吸附剂的选择性提锂机理
铝基吸附剂中Li-Al层状双氢氧化物(LiAl-LDHs)研究较多,其是由氢键、静电相互作用力以及范德华力所连接的二维氢氧化铝层状结构,由边缘共享的八面体组成,金属离子位于中心,氢氧根离子位于顶点,八面体中心位点三分之二被Al3+占据,三分之一位点被Li+占据。主体层带正电,为了电荷补偿,层间区域被水分子或阴离子填充[53],LiAl-LDHs回收Li+的示意图如图4 所示。
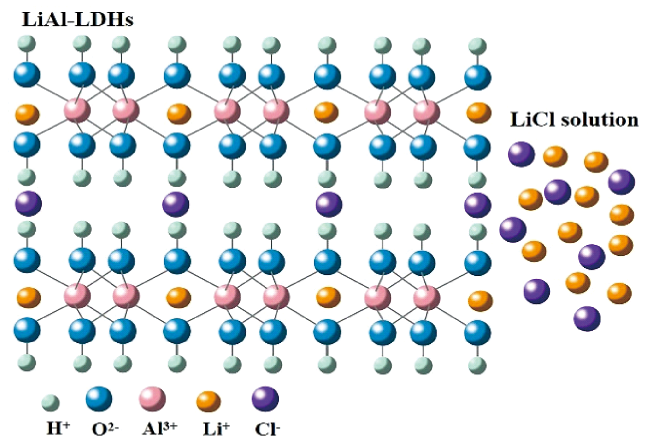
铝基吸附剂用于锂吸附之前,需要从原始结构中去除一部分Li+,以产生作为吸附位点的空位。这些空位倾向于与引入的Li+键合,形成最合适的晶体构型,从而表现出特定的Li+吸附选择性。此外,空间位阻的存在阻止了竞争性阳离子进入。所以铝基吸附剂在高Mg/Li比的盐湖中表现出良好选择性。LDHs的吸附和解吸机理可以用下式[54]表示:
$ \mathrm{LiCl}+(1-x) \mathrm{LiCl} \cdot m \mathrm{Al}(\mathrm{OH})_{3}+(n+1) \mathrm{H}_{2} \mathrm{O} \rightleftharpoons \\ \mathrm{LiCl} \cdot m \mathrm{Al}(\mathrm{OH})_{3} \cdot n \mathrm{H}_{2} \mathrm{O}+\mathrm{H}_{2} \mathrm{O}$
3.3 铝基吸附剂的选择性提锂性能
氢氧化铝基吸附剂具有制备技术成熟、稳定性和循环性优异、绿色经济等特点。相较于钛系和锰系吸附剂,Al基吸附剂具有突出的无洗脱损伤性、技术成熟度高、工业规模化运用可能性高和用去离子水就可以达到解吸效果的优点。但吸附容量以及选择性方面的不足是限制其发展的瓶颈之一。表2 比较了三类金属基吸附剂的相关性能。
表2 三种金属基吸附剂性能比较Table 2 Performance comparison of three metal-based adsorbent |
| Performance | Al-based | Mn-based | Ti-based |
|---|---|---|---|
| Li+ Adsorption Capacity | √ | √√ | √√√ |
| Li+ Selectivity | √ | √√√ | √√ |
| Technology Maturity | √√√ | √√ | √ |
| Stability and Regeneration Ability | √√√ | √ | √√ |
| Facile Operation Conditions | √√√ | √ | √√ |
| Environmental Safety | √√√ | √ | √√ |
| Low Preparation Cost | √√√ | √ | √√ |
Sun等[55]利用反应耦合分离技术从高Na/Li比盐水(Na/Li=48.7,w/w)中分离钠和锂离子,并用Li-Al层状双氢氧化物(LiAl-LDH)提取锂,Li+结合LiAl-LDH结构空位效果较好,在最佳分离条件下,锂损失低至3.93%。Chen等[56]采用分段化学共沉淀法合成了不同Fe3O4纳米颗粒含量的磁性锂铝层状双金属氢氧化物(MLDHs),MLDHs表现出优异的Li+选择性,解吸溶液的Mg/Li质量比显著降低到7.0以下。Heidari等[57]采用共沉淀法制备铝基吸附剂,氢氧化铝通过NaOH和AlCl3·6H2O以Al3+/Li+摩尔比为5加入到盐湖中制备。在温度30℃、pH=7.5时,Li+的回收率为76.4%。程鹏高等[58]通过一步沉淀法制备铝基吸附剂,并应用于泰和地下卤水提锂。结果显示,铝基锂吸附剂的平均吸附容量高达15.06 mg/g,平均脱附容量为14.11 mg/g,脱附效率为93.69%。Paranthaman等[59]报道一种从地热卤水中选择性提取氯化锂的三级台式柱萃取工艺,与Na+相比具有47.8的优异Li表观选择性,与K+相比具有212的优异Li表观选择性。对上述文献的铝基吸附剂性能总结如表3 。
表3 铝基吸附剂的合成方法与性能Table 3 Synthesis methods and properties of aluminum-based adsorbents |
| Adsorbent | Source | Method | Li+ adsorption capacity (mg/g) | pH | Selectivity (α) | Recovery rate | ref |
|---|---|---|---|---|---|---|---|
| LiAl-LDHs | AlCl3·6H2O NaOH Na2CO3 | Reaction coupling separation technology | / | / | / | 96.07% | 55 |
| MLDH (Fe3O4 doped LiAl-LDHs) | FeCl3·6H2O AlCl3·6H2O LiCl·H2O NaOH FeCl2·4H2O | Sectional chemical co-precipitation method | 5.83 | 7 | α(Li/Mg)= 362.68 | / | 56 |
| Al(OH)3 | AlCl3·6H2O NaOH brine | co-precipitation method | / | 7.5 | / | 76.4% | 57 |
| LiOH/Al(OH)3 | NaOH anhydrous aluminum chloride anhydrous lithium | single step co-precipitation | 15.06 | 6~7 | / | / | 58 |
| Li/Al-LDHs | Al(OH)3 LiOH·H2O | hydrothermal method | / | / | α(Li/Na)= 47.80 | 91% | 59 |
4 锂离子筛吸附剂
4.1 离子筛吸附剂制备
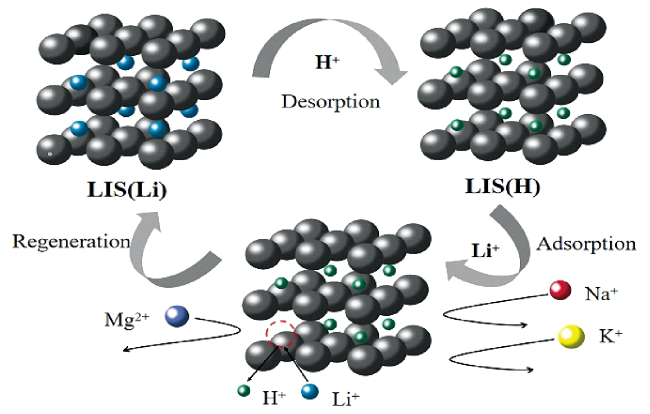
离子筛型吸附剂首先在化合物中引入Li+,经过化学合成方法制备出离子筛前驱体[LIS(Li)]。酸洗处理后,Li+从晶体结构中被洗脱出,形成锂离子筛[LIS(H)]。锂离子筛可以在富锂的溶液中选择性吸附Li+。这类吸附剂在应用过程中往往倾向于结合目标离子(Li+)形成稳定晶体构造。由于尺寸效应和空间位阻,窄的活性位点拒绝不同离子半径的其他离子占据,所以在多种杂原子共存情况下,对目标离子有特异的记忆和筛分效应。
离子筛吸附剂主要包括锰系吸附剂和钛系吸附剂。锰系吸附剂的合成与应用已经得到广泛的研究,其具有突出的吸附速率以及吸附容量,但吸/脱附过程中存在溶损问题。钛系锂离子筛的研究起步比较晚,但其具有耐酸性好、结构稳定性高、吸附容量可观等优点,应用前景开阔。
4.1.1 Li-Mn-O系离子筛吸附剂制备
1)合成LMO前躯体传统的固相合成方法包括固相煅烧以及氧化还原沉淀。石西昌等[60]利用两步固相法在煅烧温度470℃,煅烧时长6 h条件下合成前驱体Li1.6Mn1.6O4,经酸洗脱处理,材料的吸附容量为2.65 mmol/g。后续发展出了机械化学法[61]、室温固相配位法[62]等方法。机械化学法是在室温或者是较低温度下合成高度分散化合物的机械活化方法。Kosova等[61]从不同的锰(MnO2、Mn2O3、MnO)和锂(LiOH、LiOH·H2O、Li2CO3)出发,合成高度分散的系列化学计量和非化学计量的LixMn2O4尖晶石化合物。产物具有高比表面积、反应相均匀等特点。Huang等[62]首次通过室温固相配位法成功合成了LiMn2O4-yBry纳米颗粒。乙酸锂、乙酸锰、溴化锂作为原料,柠檬酸为螯合剂,将它们分别研磨成粉末,利用乙二醇400(用作分散剂)混合,后经过阶段升温制得LiMn2O4-yBry粉末。经过表征证实,LiMn2O4-yBry粉末是结晶良好的纯尖晶石相,由小而均匀的纳米颗粒组成。
2)软化学合成法也是制备离子筛常用的方法,包括溶剂-凝胶法、水热法、共沉淀法等。溶胶-凝胶法是在密封的压力容器中,以水为溶剂,在高温高压下进行反应的方法。Takada等[63]采用溶胶-凝胶法,在O2气氛中使乙酸锂(LiOAc)和硝酸锰(Mn(NO3)2)在700℃下混合1~3天,合成了结晶良好的Li4Mn5O12粉末。水热法可以通过控制水热条件,获得纳米线、纳米球、纳米片、纳米立方体等不同形貌的产品[64,65]。Zhang等[66]将水热法和低温固相反应法结合,合成b-MnO2、尖晶石型Li4Mn5O12和纯立方相MnO2纳米棒。与传统的高温煅烧工艺相比,更利于控制具有良好孔径分布和高表面积的纳米晶结构。初始锂浓度仅为5.0 mmol/L下,其吸附容量显著提高到6.62 mmol/g。此外,还有熔盐法[67]、生物分析法[68]等。
4.1.2 Li-Ti-O系离子筛吸附剂制备
文献中常报道的锂钛系离子筛通常包括Li4Ti5O12和Li2TiO3两种。它们的合成方法与锰系锂离子筛相同,包括固相烧结、水热法、溶胶-凝胶法等。Zhang等[69]采用溶胶-凝胶法合成Li2TiO3,Li2TiO3的Li+脱出率达到78.9%,钛离子的溶解率低至0.07%。Zhao等[11]通过水热法合成Li4Ti5O12纳米棒,在24 mmol/L LiCl溶液中,H4Ti5O12纳米棒的最大吸附容量达到23.20 mg/g。Li等[70]通过水热法制备了具有分级介孔结构的超薄纳米片组装Li4Ti5O12多孔微球。它们的高比表面(>180 m2/g)使之暴露出更多的吸附位点,并显著加快了吸附速率(1 h内达到平衡)。此外,H4Ti5O12微球显示出吸附容量达到43.20 mg/g。Shi等[71]采用高温固相合成法制备了Li2TiO3,Li+的提取率达98.86%,钛的溶解损失率仅为0.17%。在含694.1 mg/L锂离子的LiOH溶液中,平衡吸附容量达到39.8 mg/g。
4.2 锂离子嵌入/脱嵌机理
Li+在锂离子筛内部的插层/脱层通常由氧化还原机理、离子交换机理和复合机理来解释[72]。
4.2.1 氧化还原机理
根据Mn/Li比和晶体结构不同,锂锰系离子筛可分为LiMn2O4、Li1.33Mn1.67O4(Li4Mn5O12)和Li1.6Mn1.6O4(Li2Mn2O3)三种。前两种尖晶石基前驱体呈现立方晶体结构,Li和Mn分别占据LiMn2O4的四面体和八面体位置,Li2Mn2O5的结构模型尚未明确报道。在Hunter[73]的早期研究中,认为吸/脱附属于氧化还原过程。Li+在脱嵌的同时,Mn3+在酸性环境中发生歧化反应,转化成Mn4+和Mn2+,Mn2+溶解在溶液中造成材料损失。同时,锂离子从晶体内部扩散到表面,溶于酸性溶液中。其反应式如(1)。Ooi等[74]证实Li+吸附插入λ-MnO2四面体位点的过程中,涉及到两步Mn4+被还原为Mn3+和OH-被氧化为O2,Li+和电子迁移是相互独立的,其反应式如(2)。Li1.33Mn1.67O4和Li1.6Mn1.6O4的Mn理论上均属于+4价,在吸/脱附过程溶损相对较少,且Li元素占据比例高,理论吸附容量大。但氧化还原机理无法解释pH对Li+吸附容量的积极影响效应。
4(Li)[Mn(Ⅲ)Mn(Ⅳ)]O4+8H+ →3(□)[Mn2(Ⅳ)]O4+4Li++2Mn(Ⅱ)+4H2O
4()[Mn2(Ⅳ)]O4+4Li++4OH-→4(Li)[Mn(Ⅲ)Mn(Ⅳ)]O4+2H2O+O2
在上述方程中,( )、[]、□分别表示尖晶石结构的8a四面体位点、16d八面体位点和空腔位点。具体过程如图5 所示。

4.2.2 离子交换机理
1)尖晶石结构吸/脱嵌机理
Shen等[75]早期的一项研究中表明,H+和Li+之间的离子交换提供了Li+空位,而不是Mn离子的歧化反应。他们在10~95℃温度范围内,电解制备锰氧化物,在不同温度下呈现出不同的锰原子与质子交换行为。此外,Li+交换的固体在加热时形成尖晶石LiMn2O4,并在用稀酸处理时转化为HMn2O4。Sato等[76]利用XPS表征了Li+脱出和插入的锰氧化物尖晶石结构的表面和整体上的Mn离子的价态,发现在Li+的作用下,脱出和插入过程中Mn离子的价态保持不变。这表明无论溶液pH如何,在表面上都会发生Li+和H+离子交换反应。Koyanaka等[77]通过分析几种尖晶石型λ-MnO2的组成和吸附能力之间的相关性,验证了锂吸附能力与氢离子含量成正比。他们认为,在λ-MnO2中选择性吸附锂离子的原因并非基于所谓的离子筛效应,而是仅在锂离子和质子之间发生的合适的离子交换反应。
离子交换机理说明了尖晶石型Li4Mn5O12(Li1.33Mn1.67O4)的Li+提取过程(过程由图6 所示),如反应式(3)所示:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
根据这一机制,氧化物中的Li+能够完全被H+替代,而Mn4+和Mn3+在Li+和H+交换过程中不发生改变,并保持良好的尖晶石结构。所以质子化的锂离子筛对Li+体现出高度选择性和可逆性,这也解释pH值对吸附容量和速率造成的积极影响。但是,这一理论对于元素的溶损以及吸附容量下降还不能很好解释。
反应式很好地解释了钛基离子筛在吸/脱附过程中结构稳定、溶损小等性质。
2)层状结构吸/脱嵌机理
Chitrakar等[82]认为与尖晶石结构吸/脱嵌相比,层状结构材料的吸/脱嵌过程相对简单。由于具有只能进行Li+和H+交换的限域活性位点且结构间隙小,层状结构H2TiO3能够在存在大量竞争性离子如K+、Na+、Mg2+、Ca2+等溶液中,高效选择性吸附锂离子。H2TiO3可以再生并重新用于盐水中的锂交换,其交换过程与原始H2TiO3非常相似。
Hosogi等[83]用熔融AgNO3(573 K,5 h)处理层状化合物Li2TiO3,发现Li2TiO3与Ag[Li1/3Ti2/3]O2形貌相似,说明实验中Li2TiO3晶体骨架结构不改变。Ag+不能在(LiTi2)层发生Li+交换,但它可以在形成层状材料Ag[Li1/3Ti2/3]O2的(Li)层交换。因此,(Li)层中的Li+首先被交换以形成H[Li1/3Ti2/3]O2,随后(LiTi2)层中Li+的进一步交换以形成完全交换的相H[H1/3Ti2/3]O2。这个过程说明了在Li+和H+交换前后只发生离子交换,理论上被置换的HTi2可能变回LiTi2。
4.2.3 复合机理
虽然氧化还原机制和离子交换机制可以解释离子筛在水溶液吸/脱附过程中的许多现象,但是仍然存在局限性,因此在此基础上,研究者进一步提出了复合机理。Ooi等[84]制备了三种尖晶石型氧化锰并对其进行系列表征,发现插入位点可分为三组:氧化还原型位点、Li+特异性离子交换位点和非特异性离子交换位点。证实位点的比例根据氧化锰的制备条件而变化。并且前体中锰的氧化状态的变化与不同类型位点的形成相关。Feng等[85]发现Li+提取/插入位点可分为氧化还原型和离子交换型。通常,除非晶体中Mn(Ⅲ)的数量增加,Li+离子优先从离子交换位点提取/插入,两种位点的数量分别与三价Mn离子和Mn缺陷的量相关,且随锂锰氧化物尖晶石的制备条件(热处理温度和起始材料的Li/Mn摩尔比等)变化。
4.3 锂离子筛选择性提锂性能
离子筛型氧化物材料中原料、前驱体类型、掺杂情况、热处理方法等均会影响离子筛的晶体结构和颗粒形态,进而影响离子筛的吸附性能(吸附容量和吸附速率等)。在静态和动态吸/脱附实验中,pH值、富集锂溶液的浓度、温度等也会对优化处理后的离子筛吸附性能产生改变。最后阶段的工业化运用中吸附容量与成型工艺、材料选择等密切相关。下面主要围绕影响提锂性能的四个方面展开。
1)组成的影响
制备离子筛前驱体时,Li源、Mn源、Ti源、原料摩尔比、掺杂元素等均会影响离子筛吸附性能。Zandevakili等[86]通过正交试验研究锂盐化合物、锰盐化合物、氧化剂、煅烧温度、加热时间和Li/Mn摩尔比等6个有效参数对合成的离子筛的影响,发现尽管所有提到的参数都对锂吸收能力有显著影响,但氧化剂和Li源是最有效的因素。Gu等[87]为了抑制固态反应过程中的团聚,首次将C2H3LiO2·2H2O作为锂源合成前驱体Li2TiO3。在实际盐湖卤水中获得的HTO的分离因子α(Li/Mg)达到5441.17。Tomita 等[88] 合成LiMxMn2-xO4(M=Cu、Mg、Zn;x≤0.5),并研究了取代对晶体结构和电导率的影响。随着掺杂元素增加,Mn—O键变短,Li—O键变长。XPS研究表明,随着化合物中Mg、Zn或Cu含量的增加,Mn3+转化为Mn4+,进而使得Mn元素稳定性增强减少溶损。Tian等[89]采用软化学方法合成了掺杂镁(Ⅱ)的尖晶石锂锰氧化物(LMS),并通过从LMS中提取锂和镁制备了纳米级离子筛锰氧化物(HMS)。发现Mg掺杂提高HMS稳定性,重复使用4次后,吸附容量保持在95%以上,且24 h内可从溶液中回收高达99.2%的Li+。这些在锂离子筛结构中引入能和O形成强离子键Mg、Fe、Ni等元素,能降低在酸浸时Mn的溶出率,提高实际运用能力。
2)织构的影响
通过改变离子筛织构,如增加离子筛表面积、缩小内扩散路径长度、改变孔径通道及结构形态等均能够提升吸/脱附速率。高比表面积会存在大量吸附位点,利于Li+插入和迁出。Zhang等[65]通过水热法制备了直径为20~140nm,长度约为0.8~4 μm的尖晶石Li4Mn5O12纳米棒,其比表面积达到70 m2/g左右,5 h可到达吸附平衡。多孔和大孔结构的离子筛,可以提升Li+在离子筛中渗透、交换和平衡速率。Li等[90]合成了三维大孔介孔锂离子筛(3DM-H4Ti5O12),与其无孔对应物(1.12 mmol/g)相比,3DM- H4Ti5O12(5.51 mmol/g)显示出优越的Li+吸附性能,这归因于Li+在高度互连的多孔通道中传质阻力相对少。离子筛的形态包括纳米线、纳米球、纳米片等,不同形态的吸附剂,其吸附性能不同。Moazeni等[81]通过两步水热工艺合成纳米管形态的钛酸锂尖晶石。然后用稀酸性溶液处理制备直径约为70nm、长为2 μm的Li4Ti5O12尖晶石三元氧化物纳米管。可以回收初始浓度120 mg/L锂溶液中39.43 mg/g的Li+。韩红静等[91]合成了铝掺杂锰基离子筛H1.6(Mn1-xAlx)1.6O4,样品比表面积大呈均匀光滑的纳米片多面体形貌。在初始Li+浓度为80 mg/L溶液中吸附容量达到32.32 mg/L,吸附容量大。
3)界面结构性质的影响
根据研究表明,锂离子筛浸润性、暴露面与其吸附性能密切相关。锂离子筛的润湿性对其吸附容量有很大的影响。Li+在水溶液中常常以水合离子形式存在,具有更好的润湿能力,可以使得离子筛在卤水中与Li+更充分接触,从而实现高效离子交换。Li等[92]研究不同晶相TiO2前体(非晶、锐钛矿和金红石)所制备的H2TiO3锂离子筛(HTO)对其吸附性能的影响。锐钛矿TiO2衍生的HTO-400离子筛显示出最小的接触角19°,表明基材具有优异的亲水性,Li+可以很容易地运输并与H+交换。具有不同暴露晶面的吸附剂会影响表面脱水过程和Li+吸附行为。Zhao等[12]合成了具有主(111)面的八面体组装的纳米球Li4Ti5O12(LTO-OS)和具有主(01-1)面前驱体的纳米片组装的微球Li4Ti5O12(LTE-NS)。通过酸处理获得这些相应的H4Ti5O12吸附剂(HTO-OS和HTO-NS)。由于不同脱水过程和暴露面的作用,HTO-NS(35.5 mg/g)比HTO-OS(31.2 mg/g)具有更高的吸附容量和更快的吸附速率(平衡时间<2 h)。
4.4 成型锂离子筛吸附剂
Hong等[95]使用壳聚糖作为黏合剂将LMO造粒。制备的壳聚糖LMO颗粒是中孔材料,其孔径为6.5~30.0nm。壳聚糖LMO颗粒的稳定性从88%提高到100%。当Li+浓度为30 mg/L时,HMO粉末和壳聚糖HMO颗粒表现出相似的吸附容量10 mg/g,但造粒后吸/脱附平衡时间大幅度增加。Zhou等[96]通过简单的煅烧方法合成一种新型的Zr掺杂Ti-LIS(HZrTO)。通过环氧树脂(E-12)对HZrTO进行造粒,获得了圆柱形EP/HZrTO复合材料。成型前后Li+吸附容量下降,但显示出优异的稳定性和对Li+的高选择性。解利昕等[97]通过Li1.6Mn1.6O4与高分子树脂PVDF杂化制膜,酸性溶液中Li+提取率达到95%,Mn溶损在3.5%左右。在富锂溶液中12h达到吸附平衡且该膜对Li+有较好选择性。Ma等[98]采用聚氨酯模板法制备了具有三维互通网状结构的锂锰氧化物(LMO)泡沫,交联沥青作为载体和胶黏剂。该锂子筛在制成复合泡沫前吸附容量37.22 mg/g左右,成型后为8.83 mg/g,吸附容量下降,且提锂后沥青和离子筛结合能力变弱。
上述的部分锂离子筛性能总结如表4 和表5 。
表4 不同类型钛基离子筛合成方法与性能Table 4 Synthesis methods and properties of different titanium-based ion sieves |
| Precursor | Source | Method | Li+ adsorption Capacity (mg/g) | pH | Selectivity (α) | Cycling stability | ref |
|---|---|---|---|---|---|---|---|
| Li4Ti5O12 | TTIP LiOH·H2O | solvothermal reaction | 35.5 | 13 | / | 92.5% (Five cycles) | 12 |
| Li4Ti5O12 | Ti3AlC2 LiOH | two-step hydrothermal method | 43.20 | 12.1 | α(Li/Mg) =269.00 | 93% (Twenty cycles) | 70 |
| Li4Ti5O12 | TiO2LiOH | Soft hydrothermal method | 39.43 | / | / | / | 81 |
| Li2TiO3 | C2H3LiO2·2H2O TiO2 | high-temperature calcination | 40.16 | 10 | α(Li/Mg) =5441.17 | 98% (Five cycles) | 87 |
| 3DM-Li4Ti5O12 | CH3COOLi C12H28O4Ti | hydrothermal method low temperature calcination method | 38.24 | / | α(Li/Mg) =30.00 | 80% (Six cycles) | 90 |
| Li2TiO3 | Ti(OBu)4 Li2CO3 | solid state reaction | 34.2 | 12 | α(Li/Na) =19.96 | 90.6% (Eight cycles) | 92 |
表5 不同类型锰基离子筛合成方法与性能Table 5 Synthesis methods and properties of different manganese ion sieves |
| Precursor | Source | Method | Li+ adsorption capacity | pH | Selectivity | Cycling stability | ref |
|---|---|---|---|---|---|---|---|
| 1-D Li4Mn5O12 | MnSO4LiNO3 | hydrothermal method low-temperature solid-phase reaction | 6.62 mmol/g | / | α(Li/Mg) =599.12 | / | 66 |
| LiMg0.56Mn1.50O4 | MnCl2·4H2O Mg(NO3)2·6H2O LiOH | soft chemical method | 37.4 mg/g | 12 | / | 95% (Four cycles) | 89 |
| LiMxMn2-xO4(M=Mg,Cu and Zn) | Li2CO3 CuO ZnO MgCO3MnO | high-temperature calcination | / | / | / | / | 88 |
| Li1.6(Mn0.7Al0.3)1.6O4 | MnO2LiCl AlCl3 | hydrothermal method | 32.32 mg/L | / | / | 95% (Five cycles) | 91 |
| Li1.33Mn1.67O4 | Li2CO3MnCO3 | solid-phase synthesis method | 10.00 mg/g | / | / | / | 95 |
| Li1.6Mn1.6O4 | KMnO4LiOH | hydrothermal method Solid high temperature sintering method | 41 mg/g | / | C =474.46 | 85.37% (Five cycles) | 97 |
5 其他类型吸附剂
吸附法除上述几种吸附剂外,还有层状金属酸性盐吸附剂、Li+印迹聚合物类吸附剂、天然矿石和智能吸附材料等。层状吸附剂一般为+4价金属酸性盐。由于锂是金属元素中半径最小的,所以层间距越小越能将大直径金属排除在外,对锂的选择性就越高。+4价金属酸性盐通常包括磷酸盐和砷酸盐。砷酸钍[99]晶体结构非常紧密,仅锂离子直径与其间隙匹配,能够自由进入内部发生置换,而其他离子由于尺寸和空间位阻被阻隔在外,因此显示出高度选择性。Li+印迹聚合物是最近研究较热的吸附材料,在其制备过程中引入Li+作为模板,脱锂后配位结构(“印迹空穴”)和位置不变,从而对锂有亲和性。Liang等[100]制备了磁性碳基锂离子印迹材料(Li+-IIP-Fe3O4@C) 。Li+的最大吸附容量为22.26 mg/g。Li+-IIP-Fe3O4@C在6次吸附-脱附循环后仅降低8.8%,显示出优异的再生能力,使其对锂回收非常有用。天然矿石和碳吸附材料如活性炭,通过其表面含有的丰富含氧基团(—OH、—COOH等)吸附Li+。Park等[101]利用活性炭(AC)通过化学处理在其表面引入了官能团,提高了锂离子回收量。在5个吸附-解吸循环后表现出良好的稳定性,并指出Li+是由于正负电荷的静电作用而产生吸附。Huang等[102]以冠醚和偶氮苯衍生物的混合物为功能单体,设计了基于介孔C3N4表面的新型智能光控锂离子印迹聚合物(P-IIPs)。结果表明,UV照射导致P-IIPs的Li+解吸,而可见光照射促进它们的吸附。在可见光照射下,400 mg/L溶液的最大吸附容量达到3280.5 μmol/g。这种利用紫外灯再生的吸附剂,为锂资源的回收提供了一种绿色可行的策略。
6 结论与展望
近年来,随着锂终端产品迎来爆发式增长,在未来很长一段时间,锂电等行业对锂的需求势必持续增长,从卤水、海水等水资源中提锂将是今后获取锂资源的核心所在,并有望引领锂产业的发展。铝基吸附剂虽然选择性和吸附容量相比钛锰氧化物吸附剂较差,但其技术成熟度、工艺简单、价格低廉,第一个实现了工业化运用。有机类吸附剂易于化学耦合和改性、选择性好,但价格高、合成过程复杂、单体对锂离子络合能力受多方面影响。目前倾向于向高选择性配体和高稳定性基体相结合的提锂应用材料的方向发展。无机吸附剂合成简单、成本低,且对Li+有较好的选择性和吸附容量等优势,应用前景非常好。但锰基和钛基吸附剂都存在需要改善的问题。锰基材料易溶损、稳定性差,是限制其工业化运用的最大问题,通过负载基体材料进行成膜、造粒和发泡来缓解溶损问题并且便于回收利用,基体材料的选择非常重要。钛基材料中Ti—O高键能提高了分子结构稳定性,但存在解吸困难、固相传质慢、吸附容量相对锰基较低等问题。采用合适的材料固化锂离子筛吸附剂,元素掺杂、改变离子筛结构形态以及制备方法,解决溶损、吸附速率偏慢和实际吸附容量偏低等问题,才能够极大推进其工业化的进展。其他类型吸附剂虽不是当下主流,但也给吸附剂领域开辟新的方向。本文中冠醚、铝基、锂离子筛型和其他类吸附剂优缺点总结如表6 。实现功能扩大到实际工厂运用是所有吸附剂的最终目标,也是能源发展的必然选择。因此,未来无机金属基吸附剂开发应侧重于:1)高稳定性,能够实现长期稳定循环使用。2)经济性,能够满足绿色环保和规模化生产的实际要求。3)优化的制备工艺,对制备方法、原料、温度等进行探索,获得成本低、性能优异的吸附剂。新能源背景下[103,104,105],期待制备出高品质满足工业化应用的吸附剂,促进锂资源上下端循环利用体系建设,对未来锂资源低碳应用产生全方面影响。
表6 冠醚、铝基、锂离子筛型以及其他类型吸附剂的优缺点总结Table 6 Summary of advantages and disadvantages of crown ether, aluminum based, lithium-ion sieve type, and other types of adsorbents |
| Performance | Crown ether | Alumina-based adsorbent | Lithium ion-sieve | Others |
|---|---|---|---|---|
| Adsorption Capacity | ★★ | ★ | ★★★ | ★★ |
| Selectivity | ★★ | ★ | ★★★ | ★★ |
| Technology Maturity | ★ | ★★★ | ★★ | ★ |
| Stability and Regeneration | ★★ | ★★★ | ★ | ★★ |
| Cost | ★ | ★★★ | ★★ | ★ |