




1 引言
2 高压下的简单气体
2.1 高压下的氩气和氢气
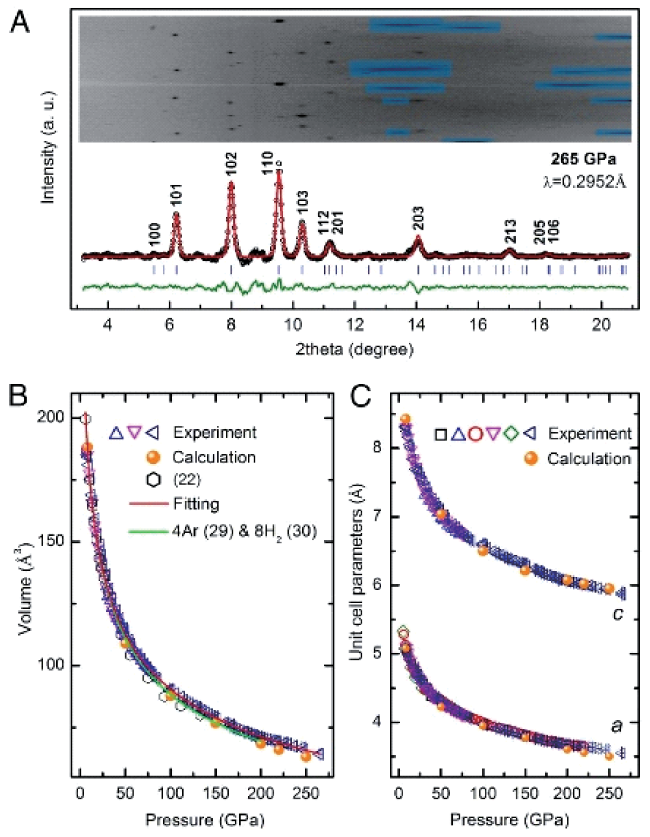
图1 (A) 在 265 GPa 下旋转 DAC ±10° 收集的X 射线衍射图的 LeBail 全谱拟合。插图为Ar(H2)2 结晶的原始图像。掩蔽区域(半透明蓝色)是来自金刚石的饱和衍射峰;(B) Ar(H2)2 的 EOS;(C) Ar(H2)2 在压力下的晶胞参数。六个样品用于确定晶胞参数a。六个样品中的三个用于确定 a、c 和晶胞体积。这是由于在压力下形成的 Ar(H2)2 晶体的优选取向[34]Fig.1 (A) LeBail full-profile fitting of XRD pattern collected with rotating DAC by ±10° at 265 GPa. (Inset) Caked raw image. Masked regions (semitransparent blue) are saturated diffraction peaks from diamond. (B) EOS of Ar(H2)2. (C) Cell parameters of Ar(H2)2 at pressures. Six samples were used to determine cell parameter a. Three of the six samples were used to determine both a, c, and unit cell volume. This is due to preferred orientations of Ar(H2)2 crystals formed under pressure[34].Copyright (2017) National Academy of Sciences, U.S.A |
2.2 高压下氙的金属化
2.3 高压下Xe-H2化合物的独特结构
2.4 高压下氙气和氟气的化学反应
3 高压下具有超导性质的气体
3.1 超导现象概述

图2 上图:超导元素固体及其实验临界温度 (Tc) 周期表。下图:超导二元氢化物周期表(0~300 GPa)。蓝色表示理论预测,红色表示实验结果[66]Fig.2 Top: Periodic table of superconducting elemental solids and their experimental critical temperature(Tc). Bottom: Periodic table of superconducting binary hydrides (0~300 GPa). Theoretical predictions indicated in blue and experimental results in red[66]. Copyright 2020, Elsevier Science Direct |
3.2 高压下预测的高温超导体
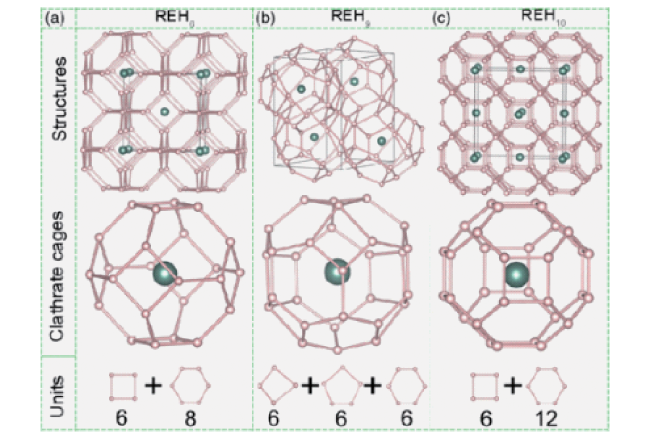
图3 REH6 (a)、REH9 (b) 和 REH10 (c) 的包合物结构。小球和大球分别代表 H 和 RE 原子。图片分别描绘了 REH6、REH9 和 REH10 的以 RE 为中心的 H24、H29 和 H32 笼。每个具有 Oh 或 D4h 对称性的 H24 或 H32 笼包含六个正方形和八个六边形或六个正方形和十二个六边形。一个 H29 笼子由6个不规则正方形、6个五边形和6个六边形组成[96]Fig.3 Clathrate structures of REH6 (a), REH9 (b), and REH10 (c). The small and large spheres represent H and RE atoms, respectively. The middle panel depicts the RE-centered H24, H29, and H32 cages of REH6, REH9, and REH10, respectively. Each H24 or H32 cage with Oh or D4h symmetry contains six squares and eight hexagons or six squares and twelve hexagons. One H29 cage consists of six irregular squares, six pentagons, and six hexagons[96]. Copyright 2017, American Physical Society |
3.3 高压下镧系多氢化物的高温超导现象
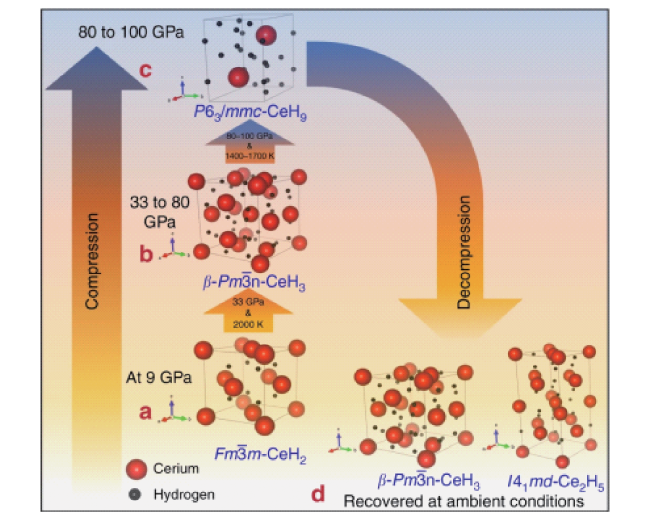
图4 各种 Ce-H 相合成和稳定性的压力温度路径。a 从 9 GPa 开始,铈与氢反应形成 ,在高达 33 GPa 时保持稳定。b 在 33 GPa 激光加热下,H2 介质中的 反应生成 在高达 80 GPa 时保持稳定。c 在 H2 介质中 80~100 GPa 的激光加热导致 超氢化物的出现。发现超氢化物相在我们研究中达到的最大压力(100 GPa)下是稳定的。d 完全减压后, 和 在环境条件下被回收[100]Fig.4 Pressure temperature path for the synthesis and stability of various Ce-H phases. a Starting at 9 GPa, cerium reacts with hydrogen to form , which remained stable up to 33 GPa. b At 33 GPa with laser heating, in H2 medium reacted to form remained stable up to 80 GPa. c Laser heating of in H2 medium at 80~100 GPa resulted in the occurrence of the superhydride. The superhydride phase was found to be stable up to the maximum pressure reached in our studies i.e. 100 GPa. d After complete decompression, and were recovered at ambient conditions[100]. Copyright 2019, Nature Communications |
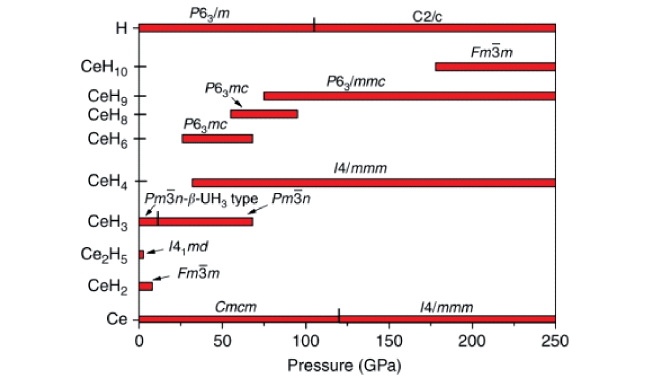
图5 理论上预测的高压下 Ce-H 系统中稳定相的压力组成相图。红色横条表示各相的稳定性范围;该相图是在进化结构预测方法 USPEX 的基础上创建的。实验发现的 P63/mmc-CeH9 预计在 78 GPa 到至少 250 GPa 时保持稳定[100]Fig.5 Pressure-composition phase diagram of theoretically predicted stable phases in the Ce-H system at high pressures. Red horizontal bars show the range of stability of each phase; this phase diagram was created on the basis of the evolutionary structure prediction method USPEX. The experimentally discovered P63/mmc-CeH9 is predicted to be stable from 78 GPa up to at least 250 GPa[100]. Copyright 2019, Nature Communications |
3.4 高压下镧系多氢化物超导的“第二岛”
4 极端高能材料
4.1 高压下的氮
4.2 高压下的氢
5 行星科学的应用
5.1 氦在行星科学研究中的应用

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
图6 在 0 K(静态条件)(黑色)、10 000 K(蓝色)、20 000 K(绿色)和 50 000 K(红色)以及沿着 ρ1/ρ0 = 4 的预压缩 Hugoniot 计算的电子能隙,其中 ρ1 是预压缩的 密度和 ρ0 = 0.1233 g·cm-3(灰色)[137]Fig.6 Calculated electronic energy gap at 0 K (static conditions) (black), 10 000 K (blue), 20 000 K (green), and 50 000 K (red) and along a precompressed Hugoniot with ρ1/ρ0 = 4, where ρ1 is the precompressed density and ρ0 = 0.1233 g·cm-3 (gray)[137]. Copyright 2008 National Academy of Sciences, U.S.A |