



1 引言
在以前的研究中,不同的色谱分析方法已被用于检测样品中的可卡因:气相色谱-火焰离子化检测器(Gas chromatography-flame ionisation detection,GC-FID)[6]、液相色谱-串联质谱法(Liquid chromatography-tandem mass spectrometry,LC-MS/MS)[7]和气相色谱-质谱(Gas chromatography-mass spectrometry,GC-MS)[8]等。色谱分析需要先在有机溶剂中提取可卡因,然后对提取物进行某种形式的净化、预浓缩和纯化,GC-MS还需要复杂的可卡因分子化学衍生程序[9]。尽管这些方法检测可卡因具有很强的可靠性和准确性,但是需要专业设备预处理样品,且从处理样品到检出结果的时间长。色谱分析的检测过程长、成本高,传统的可卡因分析方法难以满足有些场景中现场测试的需求[10]。
近年来新型的基于免疫和适配体识别元件的可卡因生物传感器已成为主流,不仅与纳米材料等信号放大的技术结合,还将新的信号转导器引入可卡因检测中。与传统的可卡因分析相比新型传感器具有特异性高、分析时间短、成本低、无需复杂的样本处理等显著优势[14]。在这篇综述中,我们系统地介绍了近年来基于免疫生物传感器和适配体生物传感器检测可卡因的各种新型策略。
2 用于可卡因检测的免疫传感器
免疫传感器的工作原理是通过抗体与抗原之间的特异性识别引起可测量的信号变化,实现对可卡因的定量检测[15]。抗体与抗原之间的免疫亲和识别促进了检测的特异性和灵敏性,使其非常适合在复杂样本中检测低浓度分子。根据标签的有无可以将免疫传感器分为标记免疫传感器和非标记免疫传感器(表1 )。
表1 不同可卡因免疫传感器及其检出限对比Table 1 A comparison of different cocaine immunosensors and their limits of detection |
| Approach of detection | Used sample | Linear detection range (mol/L) | Limit of detection (mol/L) | ref |
|---|---|---|---|---|
| Electrochemical-based ELISA | Water/Saliva/Urine | — | 4.95×10-13 | 19 |
| Colorimetric Immuno-microarray | Oral fluids | 3.63×10-9~9.9×10-7 | 3.63×10-9 | 21 |
| LFIA | Urine | 1.65×10-8~1.65×10-6 | 1.65×10-8 | 26 |
| LFIA | Saliva | 1.65×10-8~3.30×10-6 | 1.62×10-9 | 27 |
| Electrochemical | Urine/Sweat/Saliva/Serum | 1.65×10-8~8.25×10-7 | 1.19×10-8 | 30 |
| Fluorescence | PBS buffer | — | 2.30×10-11 | 31 |
| Electrochemical | PBS buffer | 0.50×10-6~2.50×10-5 | — | 34 |
| SHG | PBS buffer | — | 7.5×10-11 | 35 |
2.1 标记免疫传感器
酶联免疫吸附测定法(Enzyme-linked immunosorbent assay,ELISA)和基于纳米颗粒标记的侧向流动免疫分析法(Lateral flow immunoassay,LFIA)等技术已被广泛用于检测可卡因的免疫分析方法中。
2.1.1 酶联免疫吸附测定
Abdelshafi等[18]使用抗体IP3G2作为可卡因的识别元件,基于半抗原-辣根过氧化物酶偶联物与可卡因竞争结合酶标抗体的原理,开发出一种竞争性ELISA检测欧元纸币上可卡因的分析策略。他们通过交叉反应性研究,得到抗体IP3G2对可卡因的亲和力高于可卡因的代谢物。并且对从柏林不同地区获得的六十五欧元纸币进行了可卡因分析,检测到的可卡因的污染频率为100%。
Zhang等[21]设计了一种免疫微芯片用于体液中多种非法药物(可卡因、吗啡、苯丙胺)的定量检测。首先可卡因-牛血清蛋白复合体通过氨基与羧基的酰胺化反应固定到芯片上,复合体可以与待测物竞争结合溶液中的标记抗体。通过使用三种不同的比色反应,观察药物的不同颜色以及位置,“立即”进行药物识别。该比色免疫微芯片可以同时分析多种检测物,增强了ELISA在实际检测和现场分析方面的潜力。
2.1.2 侧向流动免疫分析法
在20世纪60年代末,LFIA作为一种低成本、快速、简单的检测方法出现,被用在即时检测(Point of care testing,POCT)的小分子检测上[22]。与ELISA相比,LFIA由于体积小易携带,信号读取方便,更适用于实验室外的现场检测。
如图1 所示,LFIA测试条通常由背垫、硝酸纤维素膜(NC膜)、样品垫、结合垫和吸收垫组成。其中背垫为检测装置提供了一定的机械支撑。NC膜为目标物检测过程中的捕获识别提供了平台,捕获抗体可以通过静电相互作用、氢键或疏水力等作用固定在NC膜上,形成测试线(T线)和控制线(C线)。结合垫是标记抗体附着的地方。吸收垫基于毛细效应提供驱动力,有助于保持液体在膜上的流动,还可以吸走多余的试剂,防止样品倒流。LFIA分为两种形式:夹心形式和竞争形式。在夹心形式中,当结合垫与目标物接触,标记抗体立即被释放,形成抗原-抗体复合物。复合物在流经修饰了捕获抗体的T线时,分析物夹在两个抗体之间形成夹心结构,T线上标签聚集,表现为阳性结果。而没有与目标物结合的标记抗体越过了T线,被C线上另一种类型的捕获分子识别并结合[23]。反之,若样品中没有目标物存在,T线上抗体就不能捕获到标记物,结果呈现阴性。在竞争形式的LFIA中,目标物与固定在T线上的半抗原竞争结合标记抗体的结合位点。当目标分析物不具有多个抗原位点时,使用竞争形式的LFIA检测[24]。基于抗原抗体的高结合效率,LFIA可以在有限的条件下对少量的样品提供定性或定量的检测[25]。
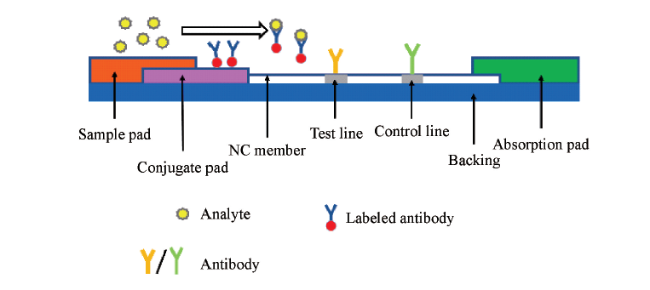
由于传统的LFIA法不容易提供对分析物的定量检测,Wu等[26]将竞争形式的LFIA与智能手机图像处理技术结合,开发了一种成本低、便捷的可卡因定量分析策略。磁珠上修饰的羧基被活化后与可卡因抗体的氨基共价结合形成(Magnetic beads-antibodies,MBs-Ab)复合物,样品中游离的可卡因(Cocaine, CC)与T线上固定的牛血清蛋白-可卡因(Bovine serum albumin-cocaine,BSA-CC)竞争结合MBs-Ab的识别位点。待测液中可卡因含量越少,T线上聚集的MBs-Ab就越多,呈现出更深的颜色。当样品溶液中没有可卡因时,MBs-Ab与T线上的BSA-CC完全反应,颜色最深。样品流过T线后,过量的MBs-Ab或MBs-Ab-CC可以与C线上固定的山羊抗鼠IgG反应,形成棕色的条带。通过安装在智能手机中的图像捕捉软件,将结合了MBs的T线和C线上的颜色强度转换为数字信号,实现对可卡因的定量检测。Ghorbanizamani等[27]选择了两性聚合物空心球包裹的罗丹明B作为抗体的标记物,使用智能手机辅助成像,实现了竞争LFIA定量分析唾液中的可卡因。由于聚合物膜的两亲性,可被用来捕获各种亲水或疏水的标记分子,使用该聚合物不仅稳定了标记分子,还促进了分子之间的结合及功能化。
2.1.3 其他免疫传感器
大多数伏安测量装置采用三电极系统:工作电极、参比电极、对电极。丝网印刷电极(Screen-printed electrode,SPE)可以将三电极系统印刷在同一基板上,将待测液滴加在SPE表面就可以覆盖多个电极,直接对待测物进行电化学检测[28],通常适用于在微量溶液中的检测。SPE技术可以用于低成本、一次性、大规模生产的伏安传感器,且具有响应快速、低功率、便捷等优点[29]。Sanli等[30]使用氧化钴作为抗体的电活性标记物,SPE为检测平台,提出了一种通过差分脉冲伏安法(Differential pulse voltammetry,DPV)实现可卡因检测的生物传感器,实现了在1.65×10-8~8.25×10-7 mol/L范围内对可卡因的定量检测。
Paul等[31]将荧光标记的抗体IP3G2与竞争形式的免疫微流体技术结合。标记抗体与待测样品混合物被注入亲和柱中,亲和柱上固定了大量的半抗原(苯甲酰芽子碱)。在可卡因存在的情况下,一些标记抗体的结合位点被阻断,亲和柱上捕获到的标记抗体数量减少,最后通过流体中荧光信号的变化分析出可卡因的含量。
2.2 非标记免疫传感器
考虑到高质量标记的抗体可能影响与目标物的识别反应动力学,导致分析物的定量不准确[32]。免疫生物传感器除了可以通过将标记固定在抗体上,通过标记抗体识别抗原来指示检测信号[33],还可以通过无标记法实现对目标物的检测。Sengel等[34]将修饰了带有电活性单体的玻碳电极作为检测平台,检测在[Fe(CN)6]3-/4-溶液中样品加入前后电信号的变化。产生这种信号变化是由于可卡因与抗体的结合阻止了通过传感器的电子转移。该传感器设计中加入的共轭聚合物技术使抗体的固定更有优势,还提高了传感器的灵敏度。Tran等[35]基于光学二次谐波(Second harmonic,SHG)技术实现了无标记免疫测定法直接检测可卡因,达到7.5×10-11 mol/L的检测限(存在干扰时)与传统的竞争性ELISA(检测限为1×10-9 mol/L)相比,该方法具有更高的灵敏性和选择性。
3 用于检测可卡因的适配体传感器
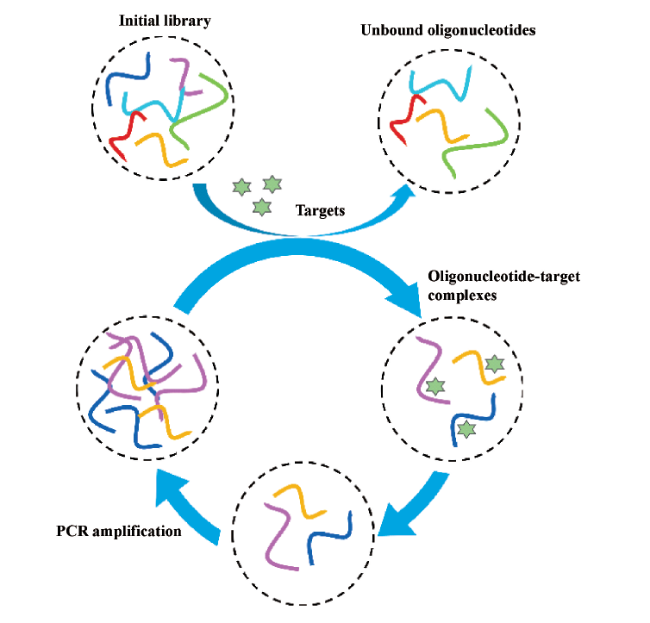
核酸适配体是非天然存在的结构化寡核苷酸,对靶标有着和抗体相似的特异性和亲和力。核酸适配体通过链内或链间的碱基配对、碱基堆叠相互作用、范德华力等自发折叠成独特的结合结构(包括二级结构和三级结构)。适配体在这些力的作用下,形成发卡、凸环等二级结构作为结合结构的基础,或再进一步折叠成紧凑的三级结构后,拥有了与目标物结合的能力[36]。这些核酸分子是在体外从随机寡核苷酸文库中被筛选出来,这个过程被称为通过指数富集的配体系统进化(Systematic evolution of ligands by exponential enrichment,SELEX)[37]。SELEX的过程包括构建寡核苷酸文库、目标物与核酸链的结合、核酸链的洗脱、扩增到文库中的迭代步骤(图2 )。初始寡核苷酸文库中含有1014~1016个单链DNA或RNA分子,这些分子包含了一个中心随机序列以及5'和3'端的两个恒定引物结合位点,便于后续的核酸链扩增。目标物在一定条件下与随机文库一起孵育,未与目标物绑定的序列被丢弃。将结合了目标物的核酸链从靶标上洗脱下来,进行扩增并构建单链池,用于下一轮的亲和力筛选。最后一轮,对最终筛选出的核酸链进行测序,并对该链与目标物结合的亲合力进行表征。
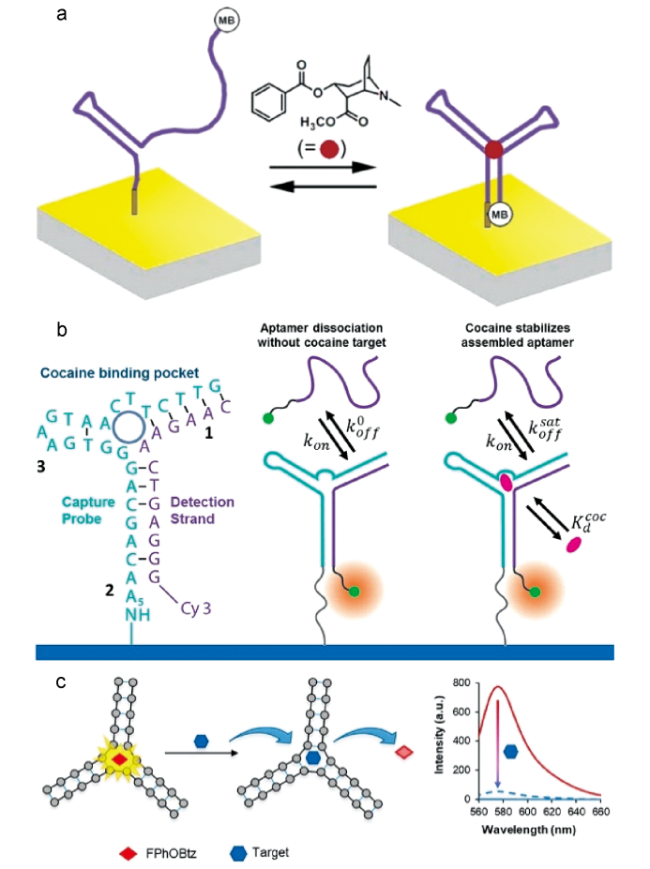
通常,适配体与可卡因的相互作用涉及一种展开-折叠机制:只有在可卡因存在的情况下,适配体才会形成三通结构(Three-way junction, 3WJ)[45]。根据适配体的片段数量大致可以将可卡因适配体分为三类。第一类单个适配体链,称为整体适配体(Monolithic aptamers,MAs)[46],其检测主要依赖于适配体的构象变化(图3a )。第二类是双片段适配体(Double-fragment aptamers,DFAs),一般是MAs被切成两段——捕获探针和报告探针[47]。由于两条链的物理分离,探针修饰的灵活性增加,为适配体传感器开发了新的检测途径(图3b )。第三类是三片段适配体(Triple-fragment aptamers,TFAs),当三个片段的适配体与可卡因同时存在时,才能形成三向的适配体-可卡因复合物[48]。与DFAs相比,TFAs有更多的开放终端可用于标记,这为探针设计和优化提供更多选择(图3c )。为了改进检测系统,可以在不影响靶分子亲和力的情况下,用报告分子对核酸适配体进行化学修饰和标记[49]。利用这三种类型的探针特异地捕获可卡因,再通过金属纳米颗粒、酶、电活性分子、荧光团、半导体量子点等标记适配体,将可卡因与适配体的识别反应转化为可检测到的信号响应[50⇓⇓~53]。
根据标记的类型和检测方法,可卡因适配体传感器主要分为两类:光学法和电化学检测可卡因。常用的光学法包括荧光、比色法、电化学发光、表面增强拉曼散射(Surface-enhanced raman spectroscopy,SERS)[54⇓⇓~57]等检测方法。可卡因结合前后适配体的构象变化导致反应体系不同的发射特性,从而根据光信号的变化分析可卡因的含量。电化学方法包括电化学阻抗谱(Electrochemical impedance spectroscopy,EIS)、方波伏安法(Square wave Voltammetry,SWV)、DPV等,通常适配体与信号转换元件(电极)耦合[58]。表2 中依据检测方法的不同提供了基于不同适配体的可卡因传感器的检出限比较。
表2 不同可卡因适配体传感器及其检出限对比Table 2 A comparison of different cocaine aptasensors and their detection limits |
| Method | Linear range (mol/L) | Detection limit (mol/L) | ref |
|---|---|---|---|
| Fluorescence | — | 5×10-6 | 62 |
| Fluorescence anisotropy | — | — | 63 |
| Fluorescence | 0~1×10-5 | 5×10-8 (in 10% saliva) | 64 |
| Fluorescence | 5×10-10~8×10-8 | 8.4×10-11 | 65 |
| Fluorescence | 0~1×10-10 | 5.4×10-13 | 66 |
| Cas-12a based fluorescence | 4.7×10-7~1.5×10-2 | 3.4×10-7 | 67 |
| EWF-based fluorescence | 1×10-5~5×10-3 | 1.05×10-5 | 68 |
| Fluorescence | 1×10-6~5×10-4 | 2.5×10-7 | 69 |
| Fluorescence | 1×10-7~1×10-4 | 4.6×10-9 | 72 |
| Fluorescence | 1×10-8~1×10-4 | 8×10-10 | 73 |
| Colorimetric | — | 8.25×10-9 mol (visual) 7.79×10-9 mol (camera) | 75 |
| Colorimetric | 2×10-10~2.5×10-8 | 9.7×10-10 | 76 |
| Colorimetric | — | 1.32×10-8 mol (visual) 1.17×10-8 mol (camera) | 77 |
| Colorimetric | 0~1×10-6 | 7.49×10-9 | 78 |
| Colorimetric | 1×10-9~1.5×10-7 | 5×10-10 | 79 |
| Colorimetric | 1×10-8~1.5×10-7 | 3.3×10-9 | 80 |
| Colorimetric | 2×10-9~1×10-7 | 4.4×10-10 | 81 |
| Colorimetric | — | 1×10-5 | 82 |
| Colorimetric | 1×10-5~5×10-3 | 5×10-5 (in urine) 2×10-4 (in sweat) | 83 |
| SWV | 5×10-8~1×10-6 and 1×10-6~3.5×10-5 | 2.1×10-8 | 86 |
| SWV | — | — | 87 |
| EIS/DPV | 3.3×10-12~3.3×10-9 | 1.29×10-12 (EIS) 2.22×10-12 (DPV) | 89 |
| EIS | 1×10-15~1×10-12 and 1×10-12~1×10-7 | 3.33×10-16 | 90 |
| EIS | 9×10-11~8.5×10-8 | 2.9×10-11 | 91 |
| DPV | 3.3×10-10~3.3×10-5 | 1×10-10 | 92 |
| SWV | 3.3×10-11~3.3×10-6 | 9×10-12 | 93 |
| DPV | 1×10-11~7×10-11 | 2.6×10-13 | 94 |
| DPV | 4×10-11~1.5×10-7 | 1.5×10-11 | 95 |
| EMPAS | 2×10-6~5×10-5 | 9×10-7 | 96 |
| EMPAS | 5×10-7~5×10-6 | 3×10-7 | 97 |
| Interfacial capacitance sensing | 1.45×10-14~1.45×10-11 | 7.8×10-15 | 98 |
| FET | — | 1×10-9 | 99 |
| Conductance change | 1×10-9~1×10-5 | 1×10-9 | 102 |
| α-HL nanopore | 5×10-8~1×10-4 | 5×10-8 | 103 |
| Personal glucometer | 1×10-8~6×10-7 | 5.2×10-9 | 104 |
| LC optical sensor | 1×10-9~1×10-5 | 1×10-9 | 106 |
| LC optical sensor | 1×10-10~1×10-5 | — | 108 |
| LPFG | 2.5×10-5~7.5×10-5 | 2.5×10-5 | 109 |
| PIERS | 5×10-9~1×10-5 | 5×10-9 | 110 |
| ECL | 1×10-10~1×10-7 | 6×10-11 | 111 |
3.1 荧光适配体传感器
荧光法具有定量分析方法灵活、反应范围宽等优点。自1852年Stokes[59]引入了“荧光”的概念以来,荧光材料的许多特性被开发并应用到化学或生物分子的检测上。
在溶液中的荧光分子旋转速率受分子的固有性质(如分子体积和形状)和环境因素(如溶液的温度和黏度)的影响[60]。由于MAs在结合可卡因后构象发生改变,这种适配体构象的变化已被应用到基于改变荧光团局部环境影响荧光发射的可卡因传感器中。
当荧光各向异性(Fluorescence anisotropy,FA)的标记荧光团与配体结合形成复合物时,荧光分子的整体尺寸增加、旋转速率降低,荧光团极化高,荧光发射特性改变[61]。Liu等[62]利用四甲基罗丹明(Tetramethylrhodamine,TMR)与G碱基结合会使TMR的局部旋转受限的特性,对比了TMR分子修饰在MAs的不同结合位点上的构象变化,以及不同结合位点对TMR的FA反应。Billet等[63]设计的可卡因传感器,在可卡因存在下,阴离子荧光染料对负电荷负载的DNA排斥,使得染料几乎独立于整个DNA旋转,表现出对局部静电势的变化敏感。根据染料各向异性的读出可以量化可卡因。
由于可卡因可以诱导DFAs片段的组合,DFAs与荧光-猝灭对结合检测可卡因的策略已被开发。Yu等[64]将串联的两个靶标结合域整合在一起,设计出一种可卡因的协同结合分裂适配体(Cooperative binding split aptamer,CBSA),即一个CBSA上包含了两个可卡因结合域。一个域上的靶向结合可以稳定CBSA的结构,促进第二个结合域与可卡因的结合,表现出更高的靶结合亲和力。他们提出两种荧光猝灭方法和CBSA结合用于可卡因的检测。一种是在适配体两个结合域之间的连接部分插入可以与荧光团2-氨基-5,6,7-三甲基-1,8萘啶(2-Amino-5,6,7-trimethyl-1,8-naphthyridine,ATMND)结合的碱基空位点。可卡因诱导两段适配体组合后,ATMND与空位点结合,ATMND的荧光被猝灭。另一种是将荧光团和猝灭剂分别修饰在两条适配体链的末端。适配体与可卡因结合后,荧光团与猝灭剂靠近,发生荧光猝灭。该传感器可以在15 min内完成10%唾液中的检测。并且相较于单一结合域的DFAs(在0.5%唾液中检测限达到30 nM),该策略的适配体热稳定性和选择性都有很大的提升。Abnous等[65]将两对荧光-猝灭基团分别修饰在可卡因DFAs的末端,根据两个荧光标记的相对荧光强度的变化实现对可卡因的检测。
Gao等[66]以金纳米颗粒修饰的二硫化钼(MoS2@AuNPs)复合材料为基底,DFAs的捕获探针通过双硫醇与金作用锚定在基底上。由于MoS2不仅有良好的生物相容性,而且有较高的荧光猝灭能力,MoS2的引入很好地解决了荧光传感器中荧光报告探针非特异性吸附以及假阳性信号的问题。而且双硫醇基团提高了捕获探针和MoS2@AuNPs的结合能力,实现了检测限5.4×10-13 mol/L的可卡因高灵敏检测。
由于可卡因与MAs的结合亲合力大于MAs与其互补序列,基于可卡因与互补DNA竞争结合MAs的可卡因荧光传感器被开发。Zhao等[67]利用CRISPR-Cas12a(Clustered regularly interspaced short palindromic repeat-associated protein)的单链DNA(ssDNA)切割酶活性,实现了在4.7×10-7~1.5×10-2 mol/L内的宽范围可卡因检测。可卡因存在时,CRISPR-Cas12a的活性被激活,荧光团与猝灭团之间的ssDNA被切割开,检测到荧光信号明显增强。该传感器在检测人血清和尿液中的可卡因方面,都与高效液相色谱(High-performance liquid chromatography,HPLC)分析结果一致。Qiu等[68]设计了一种基于MAs的消逝波光纤(Evanescent wave fiber-optic,EWF)传感器用于可卡因的检测。MAs通过生物素-链霉亲和素的相互作用锚定在磁珠上,可卡因不存在时,荧光团标记的短DNA报告探针与MAs的碱基配对被结合在磁珠上。引入可卡因之后,短DNA链被竞争释放,通过消逝波平台特异性检测到上清液中释放的荧光标记的短DNA链。
Wu等[69]用小分子物质硫黄素T(Thioflavin T,ThT)作为荧光指示剂与可卡因竞争MAs的结合域。当ThT被结合在适配体提供的空腔内会有明显荧光信号产生。可卡因的存在减少了适配体结合荧光指示剂的量,荧光信号强度减弱。该方法无需对适配体进行修饰,且检测快速,几秒钟内就可以完成混合和检测步骤。
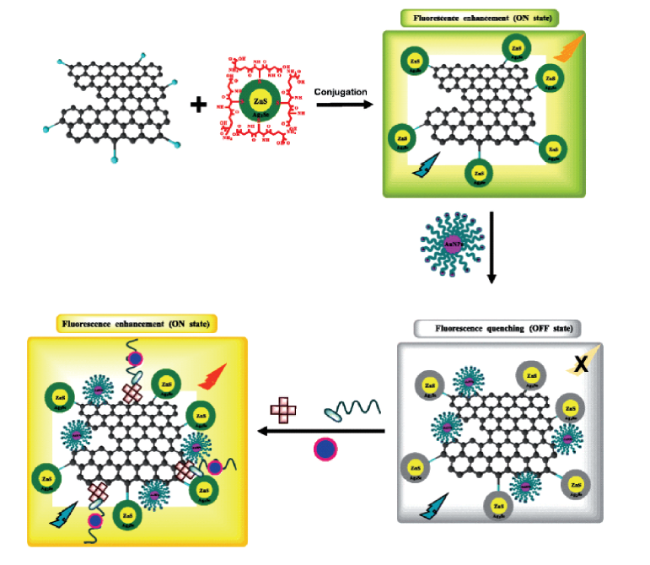
纳米材料具有独特的化学、光学、机械性质,并且可以通过静电结合、物理吸附、生物识别、共价结合的作用与不同生物分子耦合[70]。基于量子点(Quantum dots,QDs)提供荧光信号的能力,这种使用等离子体纳米颗粒的局部表面等离子体共振(Localised surface plasmon resonance,LSPR)介导的QDs荧光强度增强,实现对目标物的荧光检测的策略,已成为制造高灵敏荧光OFF-ON纳米探针的有力工具[71]。利用这个策略,Adegoke等[72]合成十六烷基三甲基溴化铵-AuNPs-石墨烯-QDs的纳米组合体,QDs的荧光被猝灭。随着可卡因被加入到反应体系中,AuNPs与QDs的纳米组合体被打开,激发了AuNPs的LSPR诱导信号,QDs荧光信号增强(图4 )。Burton等[73]同样利用等离子体纳米颗粒与QDs组合靶向诱导荧光增强的原理,使巯基化的MAs吸附在QDs-AuNPs表面,实现了对可卡因的定量检测。
3.2 比色适配体传感器
在各种分析方法中,比色传感器根据加入目标物前后颜色的不同,会有视觉或光谱的变化。由于检测结果操作简单方便、甚至可以直接肉眼观察,比色法分析可卡因的策略受到了关注[74]。
金属纳米颗粒的距离变化会影响纳米颗粒的光学特性,是比色法较为理想的标记物。由于高浓度的盐会破坏金纳米颗粒表面的电荷分布,可以起到AuNPs的聚集诱导剂的作用,使得金纳米颗粒处于聚集情况,颜色从红色变为蓝黑色。目前,已有基于AuNPs和MAs的新型生物传感器被用于可卡因的比色检测中。Wang等[75]和Sanli等[76]分别设计了在纸微流体设备和96孔板上发生盐诱导AuNPs聚集的可卡因比色传感器。由于吸附在AuNPs表面的可卡因MAs的保护,AuNPs在高浓度盐溶液中不发生聚集。但当适配体靶向结合可卡因后,适配体对AuNPs表面的保护减少,AuNPs发生聚集,并呈现出颜色的变化。之后一种四通道的纸微流体设备被开发[77],可以同时检测样品中的可卡因、可待因和甲基苯丙胺。Gao等[78]利用MoS2比表面积大、导电性好的优势,提高了AuNPs-MAs比色传感系统检测可卡因的灵敏度。在没有可卡因的情况下,适配体暂时存储在MoS2上,只有部分AuNPs不聚集。MAs与可卡因结合之后离开了MoS2,增强了AuNPs的耐盐性,有效地防止AuNPs的聚集。
Mao等[79]合成了一种镀银的纳米金颗粒(Au@AgNPs),作为传感器的比色标记物。在没有可卡因存在的情况下,可卡因MAs通过与互补链的碱基互补配对作用将Au@AgNPs与磁珠连接起来,形成了磁珠-MAs-Au@AgNPs偶联物。溶液磁性分离后,复合物被分离出来,上清液中Au@AgNPs的含量也随之减少。相反,当可卡因存在时,MAs与互补链断开,部分Au@AgNPs不再与磁珠偶联,导致磁性分离后上清液颜色比无目标物时深。通过吸光度的测量,可以得到可卡因的含量,检出限为5×10-10 mol/L。在此基础上,他们分别用AuNPs和Au@AgNPs作为标签,设计了可以同时检测两种分析物甲基苯丙胺和可卡因的比色传感器[80]。该比色法在加标的废水样品中甲基苯丙胺与可卡因的平均回收率分别为85.5%和83.9%。
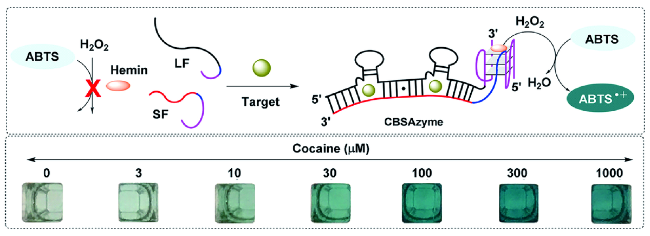
Abnous等[81]通过Au与巯基的组装,将可卡因捕获探针锚定在AuNPs表面,利用AuNPs的催化活性检测可卡因。AuNPs在TFAs-可卡因复合物的保护下失去催化活性。可卡因不存在时,AuNPs催化4-硝基苯酚被还原,样品颜色由黄色变为无色。该策略用纳米材料代替酶的催化作用,不仅规避了酶的纯化、制备及储存成本高等缺点,而且可以保持较稳定的催化活性。这对于传感器的长途运输及在恶劣环境下对目标物的检测尤为重要。同样是代替酶的催化作用,Luo等[82]利用一种新型多模块的分裂DNA结构(被称为CBSAzymes)模拟过氧化氢酶的催化作用(图5 )。CBSAzymes由两块组成,在没有靶标的时候成分离状态,靶标存在下可以有效组合,形成可以催化2,2'-联氮双(3-乙基苯并噻唑啉-6-磺酸)二铵盐氧化的络合物。该比色传感器基于CBSAzymes的检测方法,具有简单、无标记、快速、无需仪器等优点,在可卡因的现场检测方面有很大潜力。
Jing等[83]将AuNPs作为比色标记,用侧流试纸法对可卡因进行检测。利用了捕获探针-可卡因-识别探针的自发组装以及链霉亲和素与生物素之间的特异性识别,将AuNPs-DNA复合物固定在试纸条的T区和C区,显现出红色线。这种夹心式侧流试纸法可以在15 min内对尿液和汗液样品中的可卡因进行即时检测,裸眼检测限低至1×10-5 mol/L。
3.3 电化学适配体传感器
电化学适配体传感器由三部分协同工作:(1)导电平台,不仅要作为电化学反应的界面,还要为识别元件的固定提供合适的平台;(2)识别元件,电化学适配体传感器是一种以适配体为生物识别元件的电分析生物传感器;(3)信号读出设备,分析物与识别元件结合后,利用EIS、SWV、DPV等技术可以记录传感器上相应的电化学信号变化。相对于光学适配体传感器,电化学适配体传感器因其低成本、可重复使用、高灵敏度的优势在小分子快速检测中脱颖而出[84]。
已有不少研究是基于MAs与可卡因结合后构象改变引起电信号变化的电化学策略。具体来说MAs在未与可卡因结合时,处于部分折叠的较为舒展状态。与可卡因结合后,适配体处于完全折叠的紧凑状态,使适配体末端的电活性标记更靠近电极表面,从而促进电活性标记与电极之间的电子转移,引起电化学信号的变化[85]。但是这种信号变化是比较弱的,利用各种纳米材料和纳米结构(如金属纳米颗粒、共轭聚合物等)修饰导电平台实现信号放大的电化学检测技术可以进一步提升传感平台的灵敏度。Tavakkoli等[86]对金电极进行阳极氧化后用抗坏血酸作还原剂制备出纳米孔金电极,带有电活性标签的MAs通过Au—S键可以被固定在电极表面。可卡因存在时,适配体的构象变化缩短了电活性标记与电极表面之间的距离,电活性标记的电子转移效率提高,电信号发生变化。电极表面的涂覆纳米金增加了整体电极表面积和为适配体固定提供了反应平面,也降低了整体电极阻抗,提高了电极电导率和传感器灵敏度。类似的,Taylor等[87]以亚甲基蓝(Methylene blue,MB)为电活性标记物,利用植入性硅基神经记录探针实现了对体内可卡因的实时检测。通过在硅基金电极上电沉积树枝状金,增加了电极表面可用金的面积。该传感器可以在植入小鼠大脑后2 h内以高时间分辨率测量出小鼠的直接局部可卡因注射。
为了探究适配体的锚定方向是否影响传感器性能,Chamorro-Garcia等[88]以MB作为电信号标签,基于MAs构象改变研究了不同MAs适配体耦合方向对传感器电信号的影响。比较了适配体5'端和3'端锚定在电极上的传感器检测性能,发现前者表现出更大的信号增益。可能是由于3'和5'端羟基的特定几何连接的细微差异,使用5'端锚定到电极表面的DNA链比3'端锚定的更垂直,导致5'端锚定的传感器有更快的电子转移。虽然结论不具有普适性,可能与研究中适配体结构的特殊性有关,但是为优化电化学传感器的性能、提高灵敏度提供了新的思路。
除了MAs末端修饰电活性标记,还可以使用$\left[\mathrm{Fe}(\mathrm{CN})_{6}^{3-/ 4-}\right]$作探针,适配体与可卡因的结合增大了电极表面的阻抗,所以在$\left[\mathrm{Fe}(\mathrm{CN})_{6}^{3-/ 4-}\right]$溶液中可以观察到电荷转移所受到阻碍,以此实现对可卡因的定量检测。利用这个原理,Su等[89]制备出嵌入了金纳米团簇的锆基金属有机框架纳米片(AuNCs@Zr-MOF)作为传感器载体,通过Au与巯基的结合将大量的可卡因MAs链固定在该纳米片上(图6 )。可卡因结合MAs后,溶液中电活性探针$\left[\mathrm{Fe}(\mathrm{CN})_{6}^{3-/ 4-}\right]$在电极表面的电荷转移电阻发生变化。Roushani等[90]用树枝状大分子和银纳米粒子(AgNPs)对SPE表面进行改性。接着AgNPs-MAs通过与树枝状大分子上氨基的作用附着在电极表面,通过记录传感器的阻抗响应,研究电荷转移电阻值的变化。Hashemi等[91]将磁性纳米颗粒、导电聚合物与金纳米颗粒结合作为锚定适配体的传感器平台。MAs通过Au—S键固定在磁性还原氧化石墨烯/聚苯胺/金纳米颗粒复合材料上,外部磁铁可以在工作电极上捕获磁性复合材料。随着目标物-适配体复合物的形成,MAs的结构发生变化,电解液中的$\left[\mathrm{Fe}(\mathrm{CN})_{6}^{3-/ 4-}\right]$在电极表面的电荷转移电阻增加,通过阻抗响应可以对可卡因的含量进行分析。

由于氧化铟锡导电玻璃(Indium tin oxide,ITO)优异的导电性和宽电化学工作窗口以及低成本等优点被经常用于电化学传感器。Wang等[92,93]对ITO电极表面进行逐级修饰:通过3-氨丙基三乙氧基硅烷将氨基引入ITO表面,再由氨基交联剂锚定DFAs中的捕获探针到电极上。这两种传感器将活性聚合信号放大技术引入到可卡因的检测中,实现了信号放大,检测限分别达到了1.1×10-10和9.9×10-12 mol/L。Azizi等[94]将物理气相沉积技术用在ITO电极上来修饰金纳米粒子。AuNPs修饰的ITO电极比裸ITO电极具有更快的电子传输速率和更大的电流响应。DFAs中的捕获探针通过Au-S固定在电极表面,硫堇修饰的碳点作为报告探针的标记物,用以提供氧化还原信号。在最佳条件下,该可卡因生物传感器显示出1×10-11~7×10-11 mol/L的动态范围,检测限低至2.6×10-13 mol/L。
CRISPR-Cas对靶核酸序列具有超高特异性,Abnous等[95]利用末端脱氧核苷酸转移酶(Terminal deoxynucleotidyl transferase,TdT)和CRISPR-Cas12a结合的策略,实现了可卡因的高灵敏度、高选择性检测,当可卡因不存在时,适配体互补链的3'端被TdT延伸,激活了CRISPR-Cas12a的反式切割酶活性。电极表面的靶核酸链被切割,$\left[\mathrm{Fe}(\mathrm{CN})_{6}^{3-/ 4-}\right]$的电荷转移不被阻碍,导致电化学信号明显增强。
3.4 其他可卡因适配体传感器
除了电化学、荧光和比色法外,基于不同检测平台和信号产生原理检测可卡因的各种适配体传感器被开发出来,分为电学和光学两部分。
3.4.1 电学
电磁压电声学传感器(Electromagnetic piezoelectric acoustic sensor,EMPAS)提供了一个无标签传感平台,可以实时检测液体/传感器界面的质量负荷和黏弹性的变化。可卡因与适配体结合后,MAs适配体结构改变引起传感器表面的物理变化,无需标签就可以实现可卡因的定量检测。Neves等[96,97]先后报道了可卡因适配体MN4和MN6在EMPAS平台上检测可卡因。由于两种适配体的结合机制的差别发现与MN4相比,MN6(与可卡因结合时发生结构转换)的EMPAS传感器分析性能更好。Oueslati等[98]设计了一种通过检测MAs修饰的电极界面的电容变化来反映可卡因与适配体的结合状态的传感器。从测定样品到检测结果的时间为30 s,在血清中检测限为1.34×10-14 mol/L。Chen等[99]利用场效应晶体管(Field-effect transistor,FET)通过实时电信号记录观察到了适配体在结合目标物过程中的结构变化,发现该适配体与可卡因结合过程中只发生了三级结构的改变。其中石墨烯的使用在高导电性、稳定传感界面、良好的生物相容性和易于设备集成等方面提供了卓越的优势。
此外,Li等[104]提出了一种个人血糖仪检测可卡因的策略。该传感器将可卡因MAs的两条互补链分别接枝在TiO2纳米管(DNA1-TiNTA)和金纳米颗粒(DNA2-AuNP)上。适配体通过碱基互补配对作用,连接DNA1-TiNTA与DNA2-AuNP所形成的复合物,将葡萄糖分子封装到纳米管中。当可卡因存在时,可卡因与适配体反应,DNA2-AuNP与DNA1-TiNTA解离,从而导致纳米管中的葡萄糖分子被释放出来。释放的葡萄糖分子的量取决于样品中可卡因的浓度,实现了对可卡因的定量检测。
3.4.2 光学
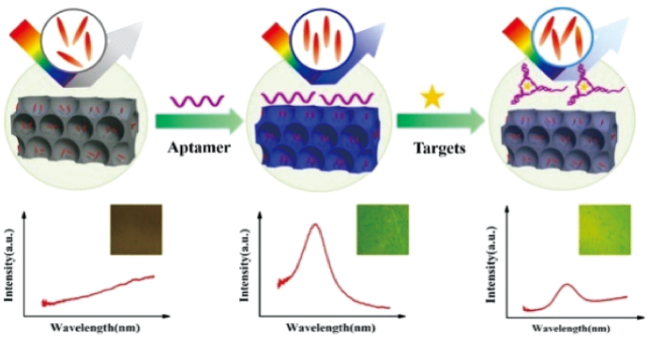
液晶(Liquid crystals,LC)是一种同时具有液体的流动性和固体的有序性的光学材料,具有与晶体相似的光学各向异性[105]。Wang等[106]使用具有两亲性的3-吗啉代丙磺酸在水/LC界面建立识别位点,用于检测可卡因。DNA介导的水/LC生物传感器的响应机制主要是DNA与其互补序列发生杂交反应或DNA构象变化引起的[107]。这种变化诱导了LC分子从垂面取向到平面取向的锚定转变。通过偏振光学显微镜可以观察到LC界面从黑色到明亮的光信号的转变。之后,Wang等[108]将LC与反蛋白石光子晶体的微孔结构相结合,构建了一种LC微阵列薄膜用于可卡因检测(图7 )。适配体结构的改变影响了微阵列中LC分子的取向,然后通过反蛋白石光子晶体的反射峰强度的变化来观察。基于适配体显著构象变化的水相/LC生物传感器具有免标记、可视化和仪器廉价等优点,是一种潜在的实时可视化检测手段。Celebanska等[109]将MN6与长周期光纤光栅(Long-period fiber rating,LPFG)相结合检测可卡因。可卡因通过引起适配体的构象重排,从而改变LPFG界面适配体层的折射率变化,产生透射光谱的偏移。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Man等[110]将AuNPs修饰在可卡因DFAs末端,构建出基于光诱导增强拉曼光谱(Photo-induced enhanced Raman spectroscopy,PIERS)效应的可卡因检测平台。通过适配体-可卡因-适配体复合物的形成,将等离子体纳米颗粒AuNPs与光激活基底(TiO2@AgNP)的距离拉近,使得分析物在紫外光诱导下获得更高的表面拉曼增强效应。
Wang等[111]将钌的衍生物用作可卡因电化学发光(Electrochemiluminescence,ECL)传感器的标记物。当可卡因存在时,MAs与可卡因结合发生结构的折叠,ECL信号物与玻碳电极之间距离减小,显示出更强的ECL特性。
4 结论与展望
在本综述中,描述了用于检测可卡因的各种免疫和适配体生物传感器,这两种可卡因生物传感器各有利弊。抗体作为识别元件对特定分析物具有很强的结合亲和力,且自然选择出来的抗体使其能完美适合于各种生物医学用途,在复杂生物样本中仍保有高灵敏度。但是对于小分子的检测,免疫传感器可能受到相似非目标干扰物的影响。相比之下,适配体合成成本更低、易于获得、结合特异性更好。但是由于核酸链的灵活性及结构不稳定性,导致在复杂环境下,适配体传感器对环境变化敏感,实验重复性较差。
除了生物识别技术,生物传感技术的进步与结合纳米技术在内的新兴技术有很大联系。纳米材料较大的比表面积为可卡因传感器提供了良好的检测平台,其在固定化、作为标记分子和换能器材料方面有着显著的优势。然而,随着纳米材料的整合也产生了新的挑战:被用于传感器检测的纳米材料需要在生理条件下保持稳定并发挥功能性,需要承受高离子强度的缓冲液,并且能在室温或体温、水溶液或环境空气中表现出良好的性能。因此,挑战在于合成和功能化这些纳米材料。需要保证做到在维持纳米材料功能性的条件下,不影响传感器的识别反应。所以聚焦于纳米材料的薄壳和聚合物的生长策略在未来可卡因传感器分析中会有较大的发展空间。
近年来,随着仿生概念的提出及发展,已有基于仿生亲和配体和分子印迹技术的化学传感器被提出用于可卡因的检测中[112⇓⇓~115]。这些可卡因捕获分子通过模仿自然识别来确定目标分析物,它们稳定性强、不易变性、对环境容忍度高,非常适合在恶劣条件下检测目标物。但是,目前基于分子探针的化学传感器仅限于实验室水平的操作和研究。基于人工捕获探针的传感器的结合亲和力及分析物选择性仍不符合实际检测的要求。并且在建立一个完整的人工分子探针之前,要准确分析仿生物的结构以及与目标物之间的结合机制。目前,人工分子探针的化学传感器研发还在起步阶段。基于人工分子探针的传感器存在的问题,已经有关于传感器与分析物之间的热力学、动力学的数据库被建立并有待拓展。
要设计出兼备稳定性、特异性、简便、抗干扰性好、可大规模生产的可卡因传感器,仍然需要付出很多努力。鉴于目前两种可卡因的特异识别元件与高灵敏检测平台以及纳米材料等技术结合的优势:简单、便携、分析快速,我们相信随着各个学科的不断交叉和进步,未来可以生产出可以用于现场检测可卡因的商业化产品。