



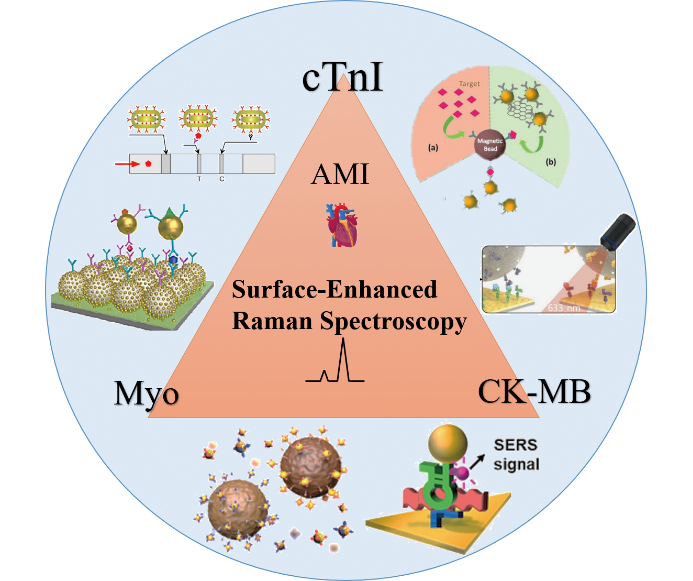
1 引言
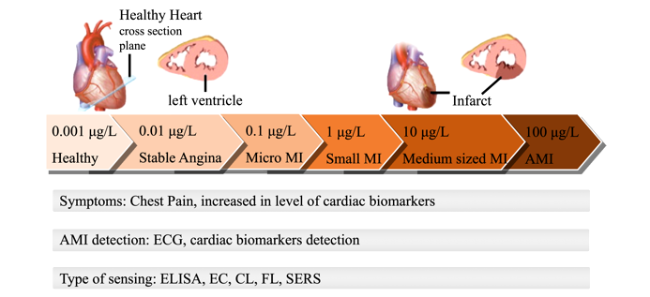
2 心肌生物标志物
2.1 心肌肌钙蛋白
2.2 肌酸激酶同工酶
2.3 肌红蛋白
3 心肌生物标志物常用检测方法及传感器件
3.1 酶联免疫吸附测定
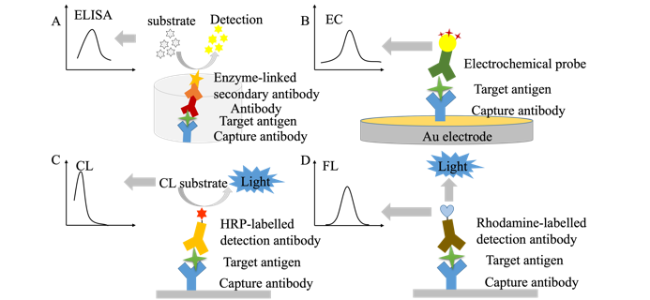
3.2 电化学免疫分析法
3.3 化学发光免疫分析法
3.4 荧光免疫分析法
4 基于SERS的心肌生物标志物检测方法
4.1 SERS及其生物传感技术

4.2 基于SERS技术的心肌生物标志物检测
4.2.1 cTnI检测
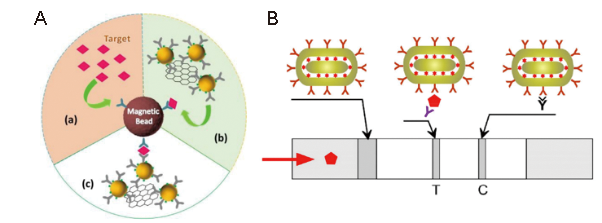
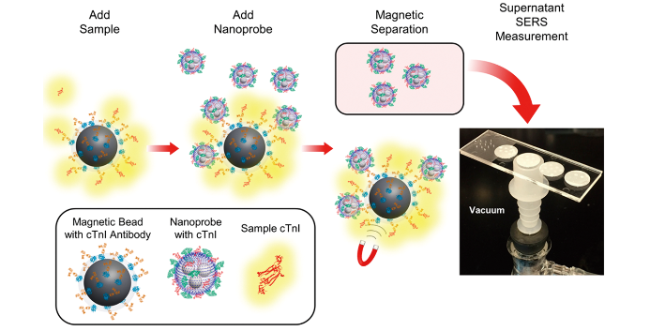
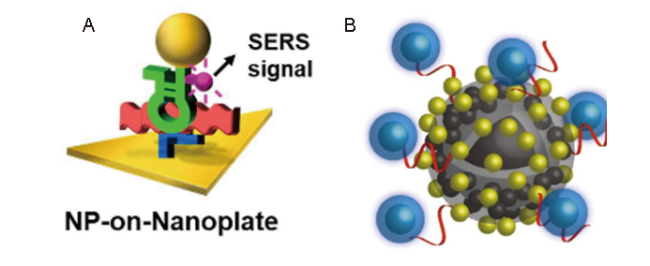
图6 (A):基于适配体识别的“Au nanoplate-cTnI-SERS适配体探针”三明治夹心结构检测原理图[120];(B):适配体标记的Fe3O4@Ag@Au定量SERS检测cTnI原理图[121]Fig. 6 (A) : "Au nanoplate-cTnI-SERS aptamer probe" sandwich structure detection of cTnI based on aptamer recognition[120]; Copyright 2020, Multidisciplinary Digital Publishing Institute; (B): Quantitative SERS detection of cTnI by Fe3O4@Ag@Au via aptamer recognition[121]. Copyright 2022, Springer |
4.2.2 CK-MB检测
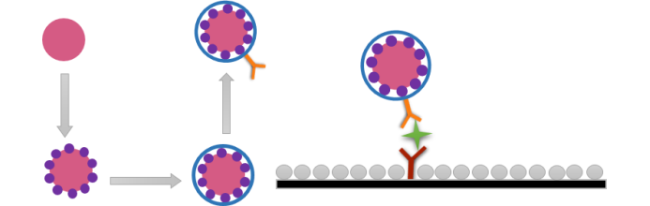
4.2.3 Myo检测
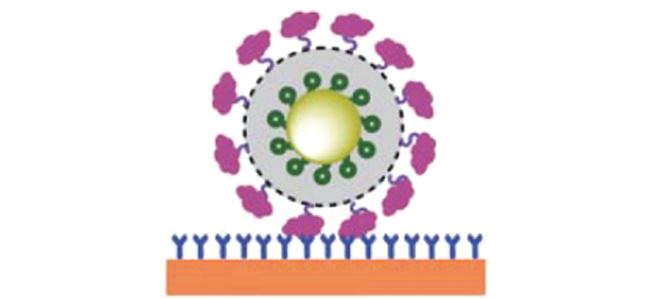
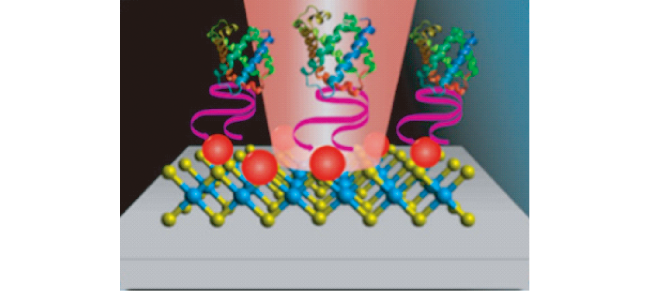
4.2.4 联合检测
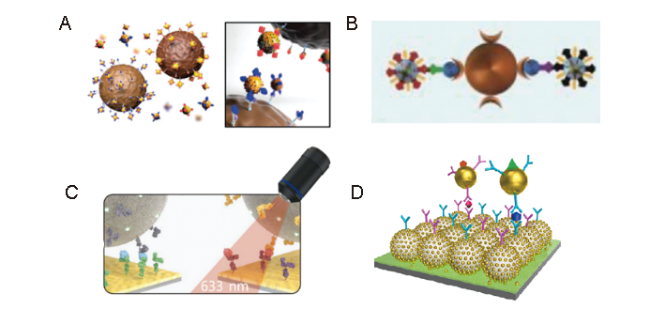
图10 (A)双元检测cTnI和CK-MB示意图[128];(B)三明治结构双元检测H-FABP和cTnI示意图[129];(C)结合金芯片的SERS免疫分析平台检测cTnI和CK-MB示意图[130];(D)基于PS微腔的SERS免疫分析平台检测cTnI和CK-MB示意图[131]Fig. 10 (A) Dual detection of cTnI and CK-MB[128]; Copyright 2013, The Royal Society of Chemistry; (B) dual detection of H-FABP and cTnI via sandwich detection structure[129]; Copyright 2020, The Royal Society of Chemistry; (C) Simultaneous detection of cTnI and CK-MB by SERS immunoassay platform combined with gold chip[130]; Copyright 2019, The Royal Society of Chemistry; (D) PS microcavity-based on SERS immunoassay platform for dual-detection of cTnI and CK-MB[131]. Copyright 2021, Elsevier |
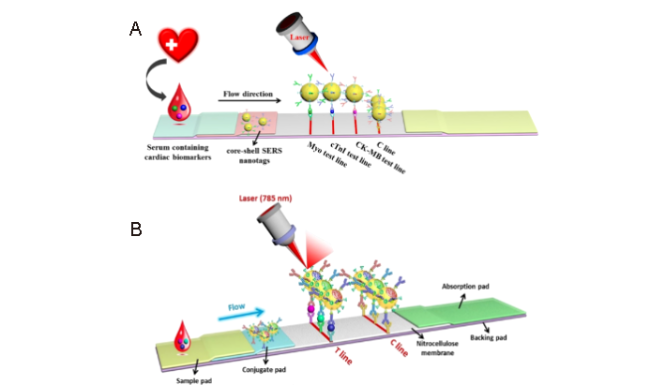
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
图11 (A):多重T线的SERS与LFIA联用方法用于同时且快速地定量检测Myo、cTnI和CK-MB[135];(B):单条T线的SERS LFIA同时检测Myo、cTnI和CK-MB[136]Fig.11 (A): Multiple T-line SERS-LFIA for simultaneous and quantitative detection of Myo, cTnI and CK-MB[135]; Copyright 2018, Elsevier; (B): Single T-line SERS-LFIA for simultaneous detection of Myo, cTnI and CK-MB[136]. Copyright 2018, Elsevier |
表1 归纳总结已报道SERS心肌生物标志物检测原理及性能指标Table 1 Summary of the reported SERS detection principles and performances on myocardial injury-related biomarkers |
| Biomarkers | Detection principles | SERS materials | Raman molecules | LODs | Linear ranges | refs |
|---|---|---|---|---|---|---|
| cTnI | Sandwich-type "capture probe(antibody functionalized magnetic bead)-cTnI-SERS immunoprobes" Combining SERS and magnetic separation | Au NPs | Malachite green isothiocyanate | 5 pg/mL | 0.01~1000 ng/mL | 116 |
| Sandwich-type "capture probe-cTnI-SERS immunoprobes" Combining SERS and LFIA | Au-Au core-shell NPs | 4-nitrobenzenthiole | 0.1 ng/mL | 0~100 ng/mL | 117 | |
| Sandwich-type "capture probe(antibody functionalized magnetic bead)-cTnI-(core-shell) SERS immunoprobes " Combining SERS and magnetic separation | Au-Ag core-shell NPs | 4-mercaptobenzoic acid | 9.80 pg/mL | 0~2.0 ng/mL | 118 | |
| Sandwich-type "capture probe (antibody functionalized magnetic bead)-cTnI-SERS immunoprobes Combination of SERS probes and immune magnetic beads PDMS enrichment device | Ag NPs | 5,5'-dithiobis(2-nitrobenzoic acid) | 3.7 pg/mL | 0~250 ng/mL | 119 | |
| Sandwich-type "aptamer-immobilized Au nanoplate-cTnI-SERS aptamer probes " Recognition of cTnI by aptamer | Au NPs | Sulfocyanine 5 | 2.4 pg/mL | 2.4 pg/mL~2.4 ng/mL | 120 | |
| Aptamers modified bimetallic magnetic nanoparticles-cTnI Combining SERS and magnetic separation and recognition of cTnI by aptamer | Fe3O4@Ag@Au NPs | Coomassie Brilliant Blue G-250 | 5.50 pg/mL | 0.01~100 ng/mL | 121 | |
| CK-MB | Sandwich-type "capture probe-cTnI-SERS immunoprobes" | gold-urchin nanoparticles | Tert-Butylhydroquinone | 10 pg/mL | 0.01~100 ng/mL | 122 |
| Myo | Sandwich-type "capture substrates-Myo-SERS probes " | Ag NPs | 4-mercaptobenzoic acid | 1.5 ng/mL | - | 125 |
| Antibody-modified substrates to capture Myo | Ag NPs | Rhodamine 6G | 10 ng/mL | 5 μg/mL~10 ng/mL | 126 | |
| Aptamer-labeled AuNP-WS2 nanohybrid capture Myo | Au NPs | Rhodamine 6G | 10 ng/mL | 10 fg/mL~0.1 μg/mL | 127 | |
| cTnI and CK-MB | Sandwich-type "capture probe (antibody functionalized magnetic bead)-cTnI and CK-MB-SERS immunoprobes Combining SERS and magnetic separation | Au NPs | Malachite green isothiocyanate and X-rhodamine-5-(and-6)-isothiocyanate | 33.7 pg/mL and 42.5 pg/mL | 10 pg/mL~1 mg/mL | 128 |
| Sandwich-type "capture probe-cTnI and CK-MB-SERS immunoprobes" | AuNPs | Malachite green isothiocyanate | 8.9 pg/mL and 9.7 pg/mL | 0 ~100 ng/mL | 130 | |
| Sandwich-type "capture probe-cTnI and CK-MB-SERS immunoprobes" Combining SERS and optical microcavity | Au NPs | 5,5'-Dithio bis-(2-nitrobenzoic acid) and 4-mercaptobenzoic acid | 3.16 pg/mL and 4.27 pg/mL | 0.01~100 ng/mL | 131 | |
| cTnI and H-FABP | Sandwich-type "capture probe(antibody functionalized magnetic bead)-cTnI and H-FABP-SERS immunoprobes Combining SERS and magnetic separation | Ag-Au core-shell NPs | 4-mercaptobenzonitrile and Thiols-poly (ethyl-ene glycol)-COOH | 639.6 pg/mL and 4.4 pg/mL | 0.0~100.0 ng/mL and 0.0~1.00 ng/mL | 129 |
| cTnI、CK-MB and Myo | Sandwich-type "capture probe-cTnI-SERS immunoprobes" Combining SERS and LFIA | Ag-Au core-shell NPs | Nile blue A | 0.44, 3.20 and 0.55 pg/mL | 0.01~50 ng/mL, 0.01~500 ng/mL and 0.02~90 ng/mL | 135 |
| Sandwich-type "capture probe-cTnI-SERS immunoprobes" Combining SERS and LFIA | Ag-Au core-shell NPs | Nile blue A, Methylene blue and Rhodamine 6G | 0.89, 4.2 and 0.93 pg/mL | 0.01~50 ng/mL, 0.01~500 ng/mL and 0.02~90 ng/mL | 136 |