



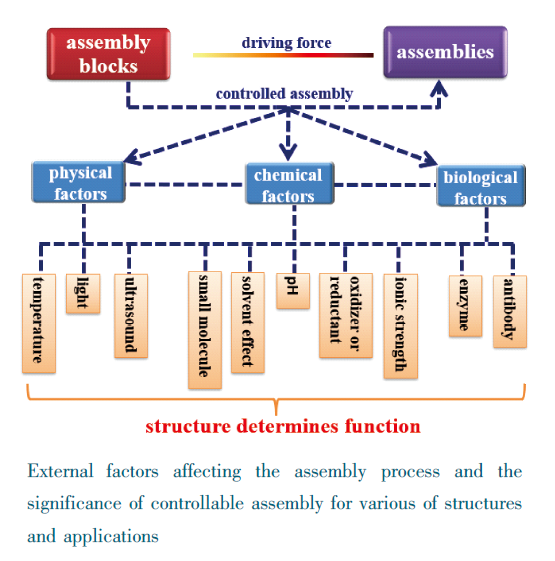
1 引言
生物体系许多重要新陈代谢过程是通过多重协同的分子间作用力自下而上的自组装过程构建的,一系列精密而灵巧的生物分子组装体结构推动了能量转化、物质传输和信息存储等各种生物功能。小到分子水平的晶体排列,大到宇宙中的星系分布,自组装过程在自然界中无处不在,如DNA的双螺旋结构,磷脂自组装形成细胞膜,蛋白质的聚集、折叠等,它们都被认为是无序的生物分子自组装的结果。受到自然界生物体的启发,研究人员开始尝试通过“自下而上”的方法来实现用原子、分子或纳米尺度功能集合体作为基元来构筑器件[1,2]。精准调控生物分子组装的动态过程,能有助于深入理解和认识进行各种生物活动的分子机制。常见的生物基元在此时就发挥了重要的作用,组装结构单元可以借助分子间弱相互作用组装而自发形成稳定的、具有特定结构和功能的、以非共价键结合的聚集体系,既可以设计得到形态仿生的结构,也可以设计成分子仿生、功能仿生的组装体,满足人们在各个领域的应用需求,自组装的方法学意义在构筑特定功能性器件等方面表现得淋漓尽致。这些生物兼容性的组装体还具有优异的力学、光学、压电和半导体性质,拓展了其在能量存储、生物传感、材料科学、生物医药、人工光合成等方面的应用研究[3,4]。
二苯丙氨酸二肽(FF)作为一种重要的生物分子,是β-淀粉样蛋白(导致阿尔兹海默症)的关键识别序列,对生命活动起到非常重要作用[5,6],被用来构筑结构多样、功能丰富的微纳米材料。FF的衍生物主要包括芳香基修饰的9-芴甲氧羰基-FF(Fmoc-FF)、萘乙酰基-FF(Nap-FF)、苄氧羰基-FF(Cbz-FF)、阳离子二肽(CDP)、二茂铁-FF(Fc-FF)以及烷基修饰的乙酰基-FF(Ac-FF)和叔丁氧羰基-FF(Boc-FF)等,这些官能团的修饰不仅丰富了FF的自组装结构,而且进一步拓展了组装体的功能和应用[7⇓~9]。为了获得功能性生物纳米材料,需要精准控制生物分子的自组装条件和过程,这也是自组装领域一直以来面临的挑战之一。前人的研究证明,通过调控分子间的相互作用(如氢键、静电作用、亲疏水作用、DNA/RNA杂化)[10,11]或改变外部条件(如调控pH、温度、离子强度或向体系中添加有机物和酶)[12⇓⇓~15]能够获得结构与功能多样的生物纳米材料,如纳米管、纳米棒、纳米球、花状、树枝状、囊泡结构等[16⇓⇓⇓⇓~21]。除此之外,还可以将一些功能化的纳米组装基元如金属纳米粒子、碳纳米管等引入自组装体系中,进一步丰富自组装生物纳米材料的功能和应用。在这个过程中,生物分子与其他非生物材料间相互作用可以指导生物分子和相应的基元自组装成杂化纳米材料[22⇓⇓~25],且组装形成各种纳米结构的机理已被广泛研究[26,27]。在这其中,分子间的相互作用、外部刺激的调控和功能的设计,对生物分子自组装形成功能性纳米材料及多功能应用意义重大[28⇓~30]。长期以来,李峻柏课题组[31⇓~33]以芳香类线性二肽-二苯丙氨酸及其衍生物作为构筑基元,通过在不同的实验条件下动态调控肽分子的自组装过程及与其他客体分子的共组装,也获得了一系列的微纳米组装体结构并开展了相关应用探究。
2 二苯丙氨酸二肽基微纳材料的可控组装
生物分子自组装形成各种微纳结构的机制相当复杂。影响其组装的内部因素是指分子间相互作用和生物分子特异性相互作用。分子间相互作用主要包括氢键、静电作用、亲疏水作用和π-π相互作用;生物分子特异性相互作用包括DNA的碱基配对、配体-受体结合、抗原抗体结合和生物分子-聚合物结合。研究人员通过改变组装条件来调控这些作用,从而实现对组装体结构和功能的精准调控。通常,改变组装条件一般包括调节内部组装环境和施加外部刺激,如温度、pH、溶剂、离子强度、酶、光照以及超声等。当然,通过选择共组装成分以及调节各组装成分之间的混合比例也是扩展组装体结构和功能的重要途径之一。
2.1 物理类因素(温度、光、超声)
2.1.1 温度
温度是影响溶液中生物分子构象和分子间相互作用的重要因素,并且该参数在各种条件下都便于精确控制。温度是调节肽基分子自组装过程的最有效也是最普遍的因素之一。Gazit课题组[34]发现FF分子本身就可以在高温条件下环化形成环肽结构,冷却沉积后线性二肽分子的组装便转变为环肽分子的组装。另外,由于大部分肽基组装材料具有温度响应性,因此可以通过改变温度实现组装体结构的转变。Liu等[35]通过冷冻处理的方法实现了FF凝胶-晶体的相转变,通过在77 K和室温之间多次循环,亚稳态的凝胶相逐渐转变为热力学稳定的晶体相。研究发现,冷冻处理破坏并重新确立了分子间相互作用力的平衡,最终得到了二肽六方晶体。该二肽晶体由于自组装体重分子间作用的振动弛豫受限,导致荧光发射显著增强。同时,实验证明这种方法可以用于其他二苯丙氨酸基有机凝胶实现凝胶-晶体转变,说明冷冻处理是一种普适化的调控生物分子组装的方法。这些发现有助于对自组装体系胶凝和晶化之间关系进行深入的研究,以达到调控单组分材料的组装和功能的目的。
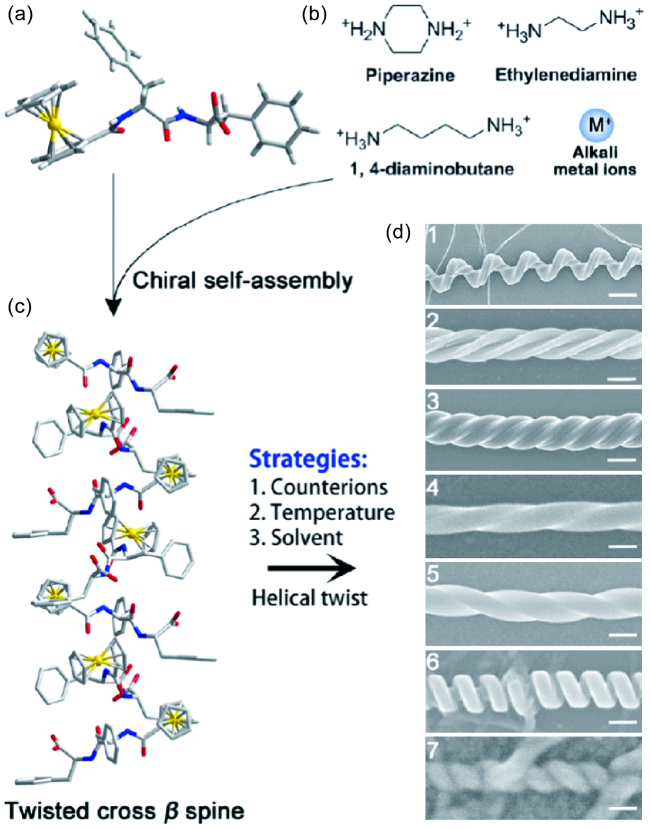
天津大学何志敏课题组[36]深入研究了Fc-FF的手性组装体系(图1 ),通过调控温度等条件可以实现对螺旋组装体直径、螺距和手性的精准调控,得到均一的手性纳米材料。组装过程中存在沿纤维轴向相互拧在一起(Twisted β-sheets)和环绕纤维轴螺旋排列(Helical β-sheets)两种结构。对手性组装行为的精确调控,可以获得理想的手性材料并探究其应用。
2.1.2 光
Ma等[37]利用一种光致异构的偶氮苯分子(3-羧基-4-羟基偶氮苯磺酸钠,EPABS)调控CDP的自组装过程,并成功实现了可见光(枝状结构)和紫外光(囊泡状结构)交替照射下组装结构的可逆转变。在可见光照射下,反式EPABS与CDP形成较强的静电相互作用和π-π相互作用,二者的芳香环发生一定程度堆叠,因此共组装形成枝状结构。当用紫外光照射时,反式EPABS转变为顺式结构,较强的亲水性以及高的空间位阻使得顺式EPABS与CDP分子之间的作用力随紫外光照射逐渐变弱甚至消失,并最终导致顺式EPABS从组装体系脱离下来从而造成共组装结构的解体。此时CDP则自组装形成囊泡状结构。
另外,Li等[40]还通过在FF自组装过程中引入光开关分子部花青(Merocyanine, MEH)实现了FF甲苯凝胶-溶胶的可逆转变,如图2 所示。在可见光照射后,MEH会向环境中释放质子,发生光致闭环反应并转变为螺吡喃(Spiropyran, SP)。FF分子中的伯胺基团接受该质子后,分子间静电相互作用增加,破坏了组装体原先的π-π堆积作用,从而导致凝胶-溶胶转变过程的发生。在黑暗环境中,SP又转化为MEH,会发生溶胶-凝胶转变。整个组装过程中,MEH分子只作为外部因素诱导组装行为发生,并不会对FF分子的组装产生影响。光刺激诱导的生物分子组装具有可逆性、快速性、无损伤、远程性和清洁性的优点,使其在光响应型生物材料、 自愈合型智能材料以及可注射生物凝胶等研究领域发挥了重要的作用。

2.1.3 超声
研究证明,超声也是促进晶体形成的非常有效的方法之一[41,42]。在晶体形成的过程中,超声的存在使分子更容易发生聚集形成非管状晶体[43]。这是因为超声处理,能够减少管状晶体形成所必须的生长晶面上的浓度梯度[44]。Skorb等[43]使用超声辅助的方法,通过调控浓度和pH分别获得了二肽纳米管和二肽纳米线结构。鉴于此,Li等[45]尝试通过改变超声时间调控FF组装体形貌。超声时间为1 min得到的是纳米纤维和微管的混合物;当超声时间增加到5 min时FF分子主要组装成微管,伴有少量的纳米纤维和微棒;当超声时间增加到20 min主要得到FF微棒结构。也就是说,超声时间的增加,有利于FF微棒的形成。随后,通过超声辅助乳液液滴模板法制备了具有单房和多房结构的空心二肽微球。甲苯和己烷与六氟异丙醇的不同溶解度、超声诱导的乳液液滴模板和GA对FF分子的部分交联是形成这两种不同类型空心球的关键因素。超声辅助的这种新型的无表面活性剂乳液液滴模板方法也有助于开发新的策略来制造具有新结构和功能的肽基微纳材料[46]。
2.2 化学类因素(外源小分子、离子强度、溶剂效应、pH)
2.2.1 外源小分子
在肽自组装研究过程中,研究者们发现仅通过单组分肽的自组装实现其在生物和相关工程领域的应用仍然存在一些困难。考虑到肽分子本身易于修饰,研究者们逐渐开始在肽自组装体系中引入功能小分子,通过不同组分之间的协同作用来实现其特殊性质的集成和优化。
Na等[47]通过将具有聚集诱导发光特性的蒽类化合物与FF共组装得到具有荧光特性的FF微管,通过改变两种组分的含量可以实现对微管形貌、荧光强度和发射波长的调控。闫学海课题组[48]发现了FF-卟啉共组装得到胶束微球结构,并且胶束微球内含丰富的水。组装过程显示,二肽首先呈 J-聚集堆积在卟啉分子周围,二肽-卟啉离子复合物再自组装得到纳米棒,这些纳米棒最后聚集得到多孔的微球。这种微球有相当宽的光谱吸收范围和较高的光稳定性,该多孔多腔的微球在光催化反应中将有巨大价值。Ma等[49]利用三种结构相似仅在端基有微小差别的磺酸基偶氮苯分子调控CDP组装得到了海胆状、花状以及平板状结构三种组装体。静电作用力和π-π相互作用是共组装结构形成的主要驱动力。实验发现磺酸基偶氮苯分子的引入破坏了CDP分子之间原有的相互作用,由于三种偶氮苯分子端基结构的微小差异,使得它们在插入CDP分子层间时与CDP的芳香环形成不同程度的重叠,因而导致了不同的分子堆积方式并最终形成了不同形貌的组装体。阳离子二肽还可以通过静电作用与磷钨酸在水溶液中共组装形成胶束,不仅对pH和温度都有良好的刺激响应性,还有很好的封装性质[50]。将八乙基铂和9,10-二苯基蒽分别作为光敏剂和猝灭剂与FF进行共组装,得到具有绿色到蓝色三线态-三线态湮灭上转换发光性能的有机凝胶。研究表明,空气中该体系上转换发光的相对量产率高达12%。同时,由于光敏剂和猝灭剂的引入增强了体系的π-π相互作用,共组装得到的有机凝胶机械性能也明显增强[51]。
FF、蛋白质及戊二醛反应形成席夫碱键,也可以进一步组装得到稳定的水凝胶。该反应条件温和,蛋白质的二级结构在组装过程中不发生改变。二肽-蛋白质水凝胶在中性PBS溶液中稳定,然而,当pH向酸性或者碱性变化时,凝胶会发生解组装,与此同时,凝胶中包封的客体分子得到释放。值得注意的是,当作为客体分子的血红蛋白(Hb)和葡萄糖氧化酶(GOD)同时封装在水凝胶中会形成 FF-Hb-GOD水凝胶,在外加葡萄糖溶液时可发生酶联反应,这使得水凝胶具有生物响应性[52]。基于以上特点,这种利用席夫碱键制备的二肽-蛋白水凝胶具有应用于生物传感器、生物催化和生物医学的巨大潜质。鉴于CDP分子的两端均含有氨基,通过小分子醛与氨基之间温和的席夫碱反应也可以调控CDP的组装。以CDP为组装基元,同样利用小醛分子诱导的共价组装方法,组装了单分散酶响应的纳米粒子[53]。除了生物小分子外,一些生物大分子(如壳聚糖、透明质酸、天然聚胺等)和肽分子的共组装体系也能够充分发挥生物大分子的功能性和肽分子组装结构的多样性,从而构建更具应用性的共组装材料。
2.2.2 离子强度
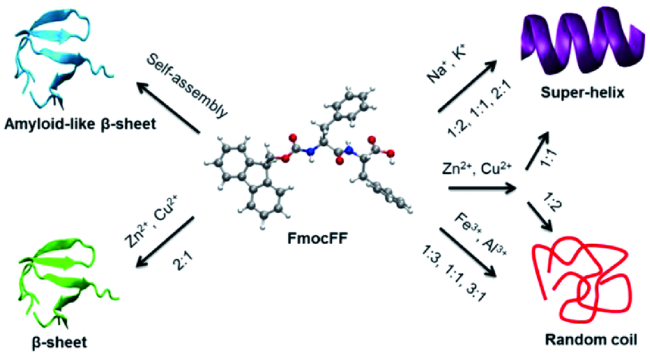
Ihee课题组[55]研究了影响形成FF纳米线和纳米管的条件。研究表明,水溶液中的离子强度对FF的组装有很大的影响。在高离子强度下,FF在水溶液中可以形成纳米线,低离子强度下形成纳米管。通过调节制备条件,FF纳米线可以分解并转变成FF纳米管。以色列科学家Gazit课题组[15]以短肽Fmoc-FF为模型,探究了金属离子在淀粉样蛋白形成过程中发挥的作用。根据金属离子的不同配位特性,他们选择了不同价态的金属离子(Na+,K+,Zn2+,Cu2+,Fe3+,Al3+),并探究了在不同比例的各种金属离子存在下Fmoc-FF的动态胶凝行为。如图3 所示,低浓度的一价金属离子形成了强度较大的凝胶,这可能是因为金属离子和Fmoc-FF的配位增强了凝胶中的超分子相互作用。然而,随着Na+/K+浓度的进一步增加又形成强度较弱的网络结构。对于二价金属离子,浓度配比也会影响组装体结构,FmocFF/Zn2+或FmocFF/Cu2+在1∶1条件下形成超螺旋结构;2∶1条件下组装形成β-折叠的结构;1∶2条件下没有自支撑的凝胶形成。整个过程中,三价金属离子参与的组装体系均未有凝胶形成。这些结果说明不同金属离子参与的肽组装机理是不一样的。另外,由于金属离子/二肽复合物带有正电荷,因此组装得到的超分子金属凝胶表现出优异的体外DNA结合和扩散能力,也为金属凝胶在3D生物芯片、细胞培养和药物输送等方面奠定基础。
2.2.3 溶剂效应
溶剂和肽分子之间的相互作用对实现自组装过程的调控至关重要。溶剂本身具有的极性、芳香性以及强氢键形成能力等性质可以直接从分子水平上影响肽分子的组装行为,因此可以通过改变溶剂得到不同的肽基组装结构。
Ihee课题组[56]对FF纳米线的形成条件和机理进行了研究。他们发现在非极性溶剂CS2中,经过常温下的超声处理后,FF能形成单分散的具有液晶行为的纳米线。这些由芳香二肽形成的纳米线具有很强的刚性,其长径比高达25.1。当纳米线的浓度为7 wt%~10 wt%时,整个分散体系表现出光的双折射性质,形成无相分离的液晶。Wang等[57]发现痕量溶剂桥连的氢键相互作用是FF自组装形成长程纤维的关键驱动力。他们选择FF/二氯甲烷作为模型,在该模型中,FF和二氯甲烷分子之间没有氢键或π-π相互作用。随后通过三种类型的痕量溶剂诱导FF组装:Ⅰ是容易和FF形成氢键相互作用的溶剂,包括乙醇、N,N-二甲基甲酰胺和丙酮;Ⅱ是与FF有π-π相互作用的溶剂,比如甲苯;Ⅲ是仅与FF具有范德华相互作用的溶剂,比如正己烷。研究表明,在体系中添加痕量易形成氢键的溶剂导致定向氢键的形成,便可以成功诱导FF自组装形成纤维或者带状结构;而添加痕量的甲苯和正己烷都不能诱导纤维结构的形成。
Zhang等[58]探究了Fc-FF在不同溶剂中的凝胶成胶能力。研究表明,水分子在Fc-FF自组装形成水凝胶的过程中发挥着至关重要的作用。相比之下,在乙腈和甲醇等极性较高的有机溶剂中,Fc-FF能够以高浓度溶解,但是不会形成水凝胶。在非极性溶剂中,Fc-FF能够通过加热溶于甲苯,但是溶液冷却后会有大量4~6 μm的小球形成沉淀析出。这可能是因为水分子与羧基和酰胺键之间形成的氢键相互作用能够驱动Fc-FF组装形成有序的β-折叠结构,这些结构进一步通过螺旋缠绕形成稳定的水凝胶。Zhu等[59]通过引入乙醇作为共溶剂触发二肽自组装体系,可以控制FF的自组装结构为有机凝胶或花状微晶。对于25%的乙醇/甲苯体系中,存在原位的凝胶相向晶体相的转变过程。溶剂的极性以及提供氢键的能力在控制凝胶的形成以及FF自组装结构中起重要作用。目前的实验结果有助于更深入地了解凝胶的形成以及凝胶剂分子在溶液中的组装过程;并且,通过简单的引入共溶剂的方法就可以实现对分子自组装结构的调控。
2.2.4 pH
众多研究表明,改变溶液pH已经成为调控肽类分子自组装的重要手段之一。肽序列中的氨基酸通常含有对pH敏感的基团,其表面电荷可以通过改变环境pH发生显著改变,这些表面电荷的变化会进一步触发静电相互作用或亲疏水平衡的变化,从而使得pH成为调节肽分子自组装行为的关键因素之一。
Ulijin等[60]报道了在纯水相中Fmoc-FF水凝胶的形成。将Fmoc-FF溶于碱性水溶液中(pH>8),用HCl滴定直到观察到有水凝胶的形成。在碱性条件下,Fmoc-FF的羧基端带负电,增大了其溶解度,并避免了由于电荷排斥导致的自组装。将体系滴定到pH<8时,部分二肽的羧基端开始质子化,使电荷间的排斥力减弱并形成氢键,利于Fmoc-FF的自组装。作者通过实验验证,提出了一个Fmoc基团悬挂的反向平行β-片层模型。4个片层叠加形成直径约为3 nm的圆柱形纤维,并通过Fmoc基团反向平行的π-π堆积来稳定此纤维结构(图4 )。这些纤维并排捆绑形成条带状结构,经过缠绕最终形成 Fmoc-FF水凝胶。

Krishnan等[61]探究了FF在不同pH条件下的自组装行为。他们发现当pH<5时,FF自组装形成35~60 nm的纳米管,随着pH的增加,这些纳米管的尺寸在几百纳米到几微米范围内变化;而在碱性pH条件下得到的组装体是由大型中空管堆积形成的网状结构。对此,他们首次提出了酸性pH条件下FF的自组装机制:即FF的等电点(pI)为5.68,低于该pH时,二肽主要以阳离子的形式存在,高于该pH时以阴离子的形式存在。 是氢键给体,而—COO-是氢键受体,—NH2和—COOH既是氢键给体也是氢键受体。在等电点之上时,由于端基—NH2和—COO-之间存在着多重氢键相互作用,FF分子通过头尾相互作用堆积。在等电点以下时,相邻— 基团之间的排斥作用限制了FF分子在水平方向上的堆积,但是垂直方向上的堆积并没有受到阻碍,因此在低于FF等电点的pH值下组装体为尺寸较小的纳米管。随着pH值的增加,— 转化为—NH2,同时增大了水平和垂直方向上的堆积相互作用,从而形成细长的大型纳米管。
Tang等[62]探究了pH对Fmoc-FF自组装行为的影响。在高pH条件下,大多数分子被离子化并且溶解在溶液中,因此没有任何组装体形成。pH在10.2~9.5时,组装体为由反平行β-折叠组成的成对原纤维。pH在9.5~6.2时,肽分子逐渐中和导致纤维表面电荷减少,从而使得它们能够通过疏水相互作用横向组装成尺寸较大的刚性带状结构。pH在6.2~5.2时,肽分子进一步中和使得带状结构发生聚集,当所有Fmoc-FF分子都被中和后,体系发生相分离,溶液中有沉淀析出。Adams课题组[63]发现2NapFF可以在高pH条件下组装形成胶束聚集体,而在低pH条件下组装形成凝胶相。研究表明,阳离子浓度的变化影响了肽分子之间的堆积方式,从而形成了不同的组装体结构。
2.3 生物类因素(酶)
徐冰课题组[68]最先进行了酶促超分子组装方面的研究,设计了一种新型的胶凝剂Nap-FFGEY,并基于激酶/磷酸化酶构建了双酶诱导体系,该双酶体系可以通过诱导胶凝剂的磷酸化和去磷酸化调节超分子水凝胶的形成[69]。在三磷酸腺苷的存在下,向水凝胶中添加激酶会引起凝胶-溶胶相变。酪氨酸残基被激酶转化为磷酸酪氨酸,使Nap-FFGEY更具亲水性;用磷酸酶处理转变得到的溶胶相便可以将其恢复为水凝胶。随后,他们将赖氨酸(K)和酪氨酸磷酸(Y)结合到Nap-FF上合成一种D型胶凝剂前体Nap-FFKY,该胶凝剂可以在磷酸化酶的诱导下组装形成水凝胶[70]。将抗癌药物紫杉醇或荧光团4-硝基-2,1,3-苯并口恶二唑连接到Nap-FFKY上,经过细胞内酶促水凝胶化在细胞内部形成荧光分子聚集体,在肿瘤化疗和细胞成像中有广阔的应用前景。Ulijn团队[71]以嗜热菌蛋白酶为酶模型诱导了多种Fmoc修饰的两亲性肽分子的自组装,发现该酶可以通过可逆水解选择性地诱导水凝胶的形成,对所得到的Fmoc-FFF凝胶进行分析发现该凝胶由直径约10~20 nm的纤维交织构成。另外,他们还利用枯草芽孢杆菌蛋白酶/嗜热菌蛋白酶双酶体系成功驱动了肽基水凝胶的自组装与水解[72]。
3 二苯丙氨酸二肽基组装体的应用
肽基组装体微纳材料因其制备简单、易修饰、生物兼容性好等优点而得到了广泛的关注和研究。超分子化学的发展、非共价键特有的动态性及方向性,进一步推动了肽基材料组装的可调性及智能响应性[73]。此部分主要介绍肽基超分子组装微纳材料在生物医学、生物传感、光电材料、光波导和催化等领域的应用。
3.1 生物医学
对健康、幸福生活的追求与向往一直都是人们生活的目标。现代医学已经进入分子医学时代,精准诊断与靶向治疗是当代医学发展的必然趋势。分子医学的发展尚处于前期阶段,面临着许多瓶颈问题,需要分子科学家尤其是化学家的深度参与。多肽优越的生物、化学性能展现了其自组装体的巨大优势和在生物医药领域广阔的应用前景。
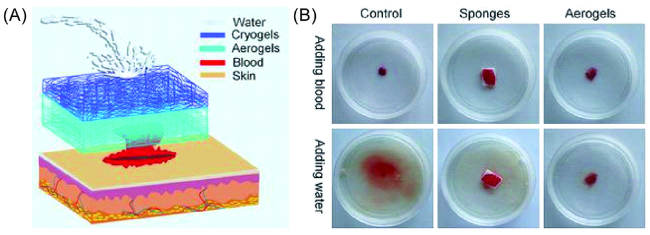
苯丙氨酸基材料具有良好的生物相容性使其能够广泛应用在生物医药领域,为人类健康服务。李峻柏课题组[74⇓~76]也一直致力于多肽组装及其生物应用方面的探索,并取得了众多的研究成果,还对基于二肽组装行为及应用进展做了比较全面的综述。Zhang等[53]研究发现,戊二醛分子(GA)能够诱导阳离子二苯丙氨酸(CDP)发生共价组装,得到单分散尺寸均一的纳米球粒子(CDPNPs)。这种纳米颗粒可以负载抗癌药物阿霉素(DOX),装载效率达到了50%以上,体外释放实验表明,DOX在含胰酶的PBS中表现了非常好的缓释性能。最近,通过精确控制二肽组装路径,利用溶剂分子诱导二肽分子取向排列,选择性地将其亲水基团或疏水基团暴露在组装的纳米结构外表面,从而形成了具有超亲水的气凝胶或超疏水的冰凝胶(将通过冷冻干燥获得的凝胶定义为冰凝胶),二者分别由纳米纤维和纳米带构成,实现了组装体表面亲/疏水性的调控,获得了界面浸润性可调控的二肽气凝胶。研究表明,所构筑的二肽组装体的性质(包括荧光发射和比表面积)也具有显著差异。进一步实验证实,上述润湿性可调控的二肽气凝胶在医药领域有其特殊的用途,将其用于体外止血,发现其具有比市售明胶海绵更好的止血效果(图5 )。该研究为发展由同一分子单元构建具有不同性质和功能的肽基组装材料开辟了新途径,同时为单组分、多性能的超分子组装材料的研究和应用带来了新的启发[77]。
光治疗因其安全性和较高的选择性被认为是一种有效的癌症治疗方法之一。通过调控肽分子间相互作用力可实现光能的定向转化,而且肽基组装材料有效地实现了一种或多种光动力药物的装载及运输,使其在光动力肿瘤治疗(PDT)及光热肿瘤治疗(PTT)领域得到了广泛而深入的研究。Ma等[78]使用交联剂戊二醛与CDP组装,得到了尺寸及结构可控的可注射型纳米载体,用于装载光敏药物Ce6及其肿瘤递送。抗肿瘤功效分析表明,包裹在水凝胶中的Ce6能够比游离的Ce6更有效地抑制肿瘤的生长和复发,而且对重要器官的损害也很小。因此,该可注射复合水凝胶有望用于浅表肿瘤的治疗。近期,又开发了pH响应可转化的肽基纳米粒子用于光动力疗法,延长肿瘤滞留时间。当暴露于酸性肿瘤微环境时,自组装肽-卟啉纳米粒子转化为纳米纤维,且这种转化提高了它们的单线态氧产生能力,并能够在肿瘤部位进行高积累和长期保留。在给药长达7天后,在肿瘤组织中检测到这些纳米材料的强荧光信号。此外,肽组装体通过体内PDT表现出优异的抗肿瘤功效。这种原位纤维转化策略可设计用于长期成像和治疗的有效刺激响应生物材料[79]。戊二醛可通过自身的羟醛缩合反应生成寡聚体,之后寡聚体的醛基与CDP的氨基通过席夫碱作用形成组装单元,静置后形成纳米粒子,其具有非常好的生物相容性和生物可降解性。通过细胞的吞噬作用,这些纳米粒子可以被细胞摄取,最终在细胞内部被降解。这种纳米粒子能很好地装载抗肿瘤药物阿霉素,以药物浓度作为衡量标准,装载之后的纳米粒子,即使药物浓度非常低时,都表现出非常好的抗肿瘤性能[53]。
3.2 生物传感
通过可控的方法组装,可以得到基于二苯丙氨酸及其衍生物的不同结构的纳米材料,因其具有比表面积大、生物兼容性好和电子传输效率高等特点,已被广泛用于电化学生物传感领域[74]。Gazit课题组[80]首次将肽基纳米管修饰在电极上并提高了电化学检测灵敏度。修饰过FF纳米管的石墨电极检测灵敏度提高了一倍;巯基修饰的二肽纳米管固定在金电极上对10 mM H2O2的响应值是裸电极的16倍,这表明FF纳米管能够显著提高电子传输效率。后来又构建了一种新型电化学酶传感器,即将葡萄糖氧化酶或乙醇脱氢酶固定在二肽纳米管上,利用示差脉冲伏安法分别实现了葡萄糖和乙醇的高灵敏检测[81]。随后,相继报道了许多肽基纳米材料用于构筑电化学性能提高的电化学传感器[82,83]。Gong等[84]将FF-Au纳米颗粒、辣根过氧化物酶先后修饰在电极上,利用该材料修饰电极对H2O2的还原性及催化活性实现了H2O2的检测。此外,也有关于苯丙氨酸二肽纳米线的电化学检测报道[85]。Castillo等[86]将叶酸修饰的二肽纳米管滴涂到石墨烯修饰的电极上实现对癌细胞的早期诊断检测。
目前已有许多相关报道证明了二肽基自组装结构材料在提高生物传感器的性能方面的应用。尽管也取得了一定的进展,但基于FF组装材料的弱导电性也限制了所构建的生物传感器的检测性能。贵金属纳米粒子具有很多突出的优点,优异的导电性、生物相容性和大比表面积等。所以,许多纳米材料修饰后的电极其电化学性能得到大幅提高。因此,将二肽分子与贵金属金纳米粒子相结合得到优势互补的材料,在提高二肽微纳组装体在生物传感器中的应用也具有很大优势[87]。最近,Wang等[88]以CDP为组装基元,利用正负电荷的相互作用,获得了尺寸可控且具有生物活性和催化性能的二肽-金颗粒杂化微球(CDP-AuNPs),将其和胆固醇氧化酶修饰在电极上,得到可用于高灵敏检测胆固醇的生物传感器(图6 )。这种复合体系很好地体现了肽的生物相容性和金颗粒的催化性能。与没有修饰CDP-AuNPs杂化微球的电极相比,CDP-AuNPs修饰后的酶电极对胆固醇检测的灵敏度提高了13倍。这种组装的颗粒也可以用于其他高度敏感和稳定性的电化学检测。
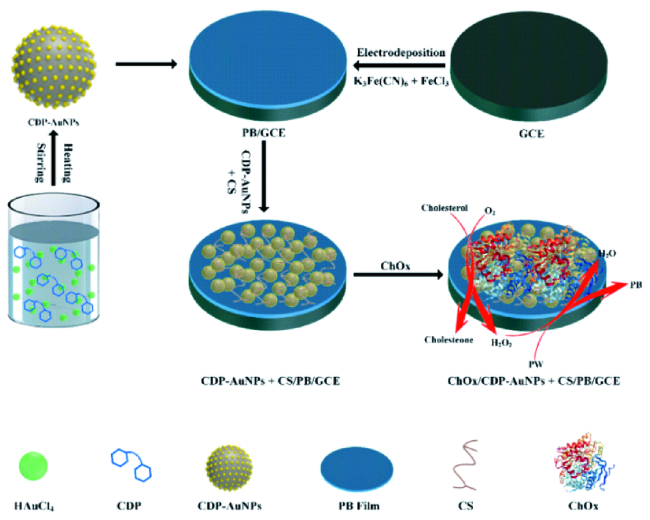
除了贵金属纳米粒子,石墨烯也可以与肽基材料共组装得到可用于生物传感的性能优异的材料。石墨烯作为一种比其他碳基纳米材料具有更高的导电性、更大的比表面积的材料,在电化学传感方面的应用前景也备受瞩目。将石墨烯与FF共组装得到的功能化纳米材料,二者性质上优势互补,既可以避免单一组分的石墨烯材料在电化学传感方面应用时存在的局限性,如在水溶液中趋向于聚集,电化学活性较弱和生物兼容性较差等[89],又可以避免单一组分的、弱导电性FF在电化学传感方面进一步应用时存在的局限性。
3.3 光电材料
随着人们对环境友好型有机半导体材料需求的日益增长,受生物启发的肽基半导体材料逐渐成为该领域中备受关注的热点。近年来,FF及其衍生物被大量应用于光电材料的构建,其所具有的光致发光性、导电性、压电性及储能性质扩展了应用范围。
Lee等[90]通过采用提拉法,控制溶剂水与六氟异丙醇的比例、FF浓度以及提拉速度,得到了同向密堆积的FF纳米管(图7 )。FF纳米管表现出压电特性以及单向极化性。借助于压电能量收集器,FF纳米管的压电电压可高达2.8 V,电流为37.4 nA,功率为8.2 nW,这一功率足以为多个液晶显示板供电。Yang课题组[91]通过电场控制,组装出垂直生长的二苯丙氨酸微棒阵列,进一步制备出二苯丙氨酸材料的微型压电发电机。该发电机的开路电压达到1.4 V,能量密度达到3.3 nW/cm2。这种微型肽基压电设备,稳定性非常高,半小时内重复按压1000次,输出电压没有降低。Heo等[92]通过气相沉积法在镀有金涂层的二氧化硅基底上沉积得到垂直排列的Cyclo-FF的纳米线阵列。这种纳米线阵列在水中及TBS缓冲液中都能够稳定存在,这种特殊的稳定性使其成功应用在摩擦产电领域。在与镀有铝涂层的PTFE接触的过程中可产生350 V的电压,输出电流达到10 μA,最大功率密度为~73.7 mW/m2,这足以点亮一百多个LED灯泡。这一研究为生物相容性能源材料的开发提供了机遇。Park课题组[93]还报道了通过肽自组装得到FF/Co3O4纳米线。肽纳米线通过高温下苯胺蒸气处理FF膜得到,再与NaBH4还原水溶液中的Co离子得到的Co3O4纳米晶混合,最终形成复合纳米线。这种纳米线可以用作Li离子电池的负极,对Li离子的储存能力远高于纯FF纳米线,未来在能量储存领域将有可能得到广泛应用。

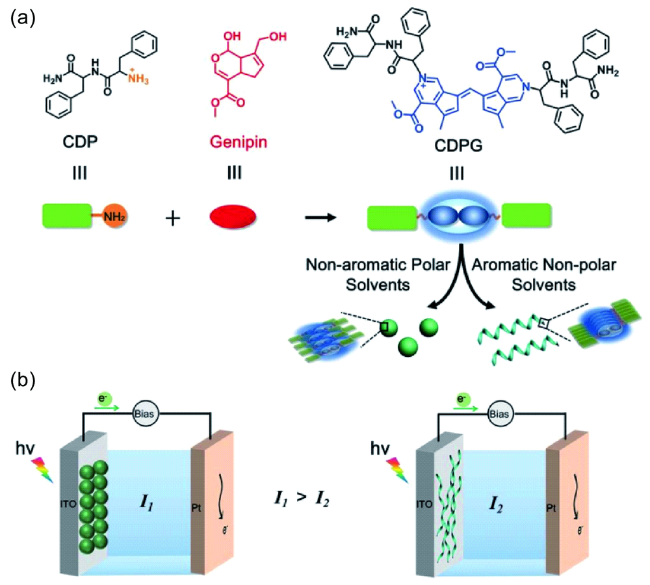
Xue等[94]利用CDP与京尼平的共价反应,获得了π共轭程度高的杂环分子(CDPG),如图8 所示。在非芳香性极性溶剂和芳香性非极性溶剂中,CDPG分别组装形成纳米球和纳米纤维。与构筑基元CDP和京尼平相比,两种组装体都表现出显著增强的光电响应效应。该光电转换器件性能增强了约500倍。实验结果表明,通过调控组装过程可以改变CDPG组装体结构达到提高器件性能的目的。
3.4 光波导
利用水热退火法控制FF缓慢结晶,可以得到高度对称的六方微米管结构。稀释六方微米管分散液,发现微米管从一端开裂,裂解成一根根独立的纳米管。这种纳米管堆积成微米管的组装模式,也与FF单晶结构信息相吻合。掺杂染料之后,FF微米管展现出良好的光波导特性,这在苯丙氨酸基组装体系中首次发现[97]。
随后,利用超声辅助下的溶剂结晶的方法,通过调控及优化组装条件(诸如超声时间、浓度、溶剂体积比),分别制备得到了二苯丙氨酸纳米纤维、微管和微棒。选区电子衍射表明制备出的二苯丙氨酸微管和微棒具有单晶结构,并表现出显著的光波导性能,这在生物分子组装体系中也是比较少见。结合肽基组装体良好的生物相容性特点,二苯丙氨酸二肽一维晶体结构有望应用在疾病诊断等医学领域[45]。
另外,还可以选取二元溶剂氨水作为自组装体系的溶剂,研究FF的组装及其光波导应用。溶液在三相接触线附近的挥发诱导了FF晶体的成核,而对基底的连续提拉体系允许晶体持续定向的生长。通过控制基底的温度、溶剂和基底的提拉速度,实现了对定向排列的FF单晶在数量和长度上的调控。不仅实现了超长定向FF单晶的快速生长,还获得了具有光波导性质的FF单晶,基于其有序排列结构,有望在大尺寸光学器件方面开展相关的应用[29]。
2019年,Li等[98]通过潮湿气流辅助下的快速挥发溶剂的方法,获得了等级有序的FF枝化结构,如图9 所示。在组装过程中引入染料罗丹明B(RhB),仍然可以获得FF枝化结构,掺杂染料的组装体在不同激发波长下均能显示出荧光,这表明FF枝化结构可以作为功能性荧光染料分子载体支架,用于生物医学检测领域。光波导实验表明,FF枝化结构表现出了单点输入、多点输出、多点输入、单点输出的光学传导模式,这在光传输器件领域有巨大的应用潜力。
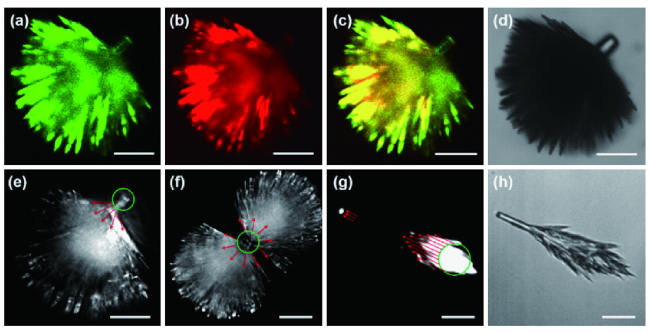
图9 (a~c) 罗丹明B掺杂的二苯丙氨酸(FF)枝化结构激光共聚焦图像及(d)明场图像; FF-RhB枝化结构的(e~g)光波导照片和(h)明场照片,标尺为 20 μm[98]Fig. 9 (a~c) CLSM images and (d) bright-field image of rhodamine B doped diphenylalanine branched structure. (e, f) Photoluminescence images of rhodamine B-doped diphenylalanine branched structures excited at the rod position. (g) Photoluminescence image and (h) bright-field image of rhodamine B-doped diphenylalanine branched structures excited at the tips of the branches. All the scale bars represent 20 μm[98] |
3.5 催化
自然系统通常具有巨大的分子复杂性,并通过精确的空间和时间控制构建精美的架构和功能材料,肽和蛋白质的超分子自组装是关键因素之一[99,100]。在多种天然酶系统中,它们优异的特性和功能不仅吸引人们去解开自然的奥秘,而且在绿色生活设计先进材料方面也引起了人们极大的兴趣。然而,天然酶具有固有的不稳定性和复杂性,因此希望开发出天然酶的仿生催化替代品。就这一目的而言,科学家们已经通过与多种非共价相互作用相关的自组装策略进行了超分子纳米酶的开发。肽作为天然蛋白质的功能基序,可作为设计功能仿生纳米酶的通用且有前景的构建单元。重要的是,基于肽的纳米酶结构调节的机制明确,以及它们作为生命系统中基本生物单元的独特性质(如选择性、自催化等)可能为调节催化性能提供进一步的可能性。Han等[101]综述了基于肽的超分子纳米酶的研究进展,包括基于肽的组装、通过超分子非共价相互作用进行的精确功能调节、催化反应和能量转换机制。这些先前的研究可以启发构建一种在各种应用中具有高催化选择性和效率的新型超分子纳米酶。
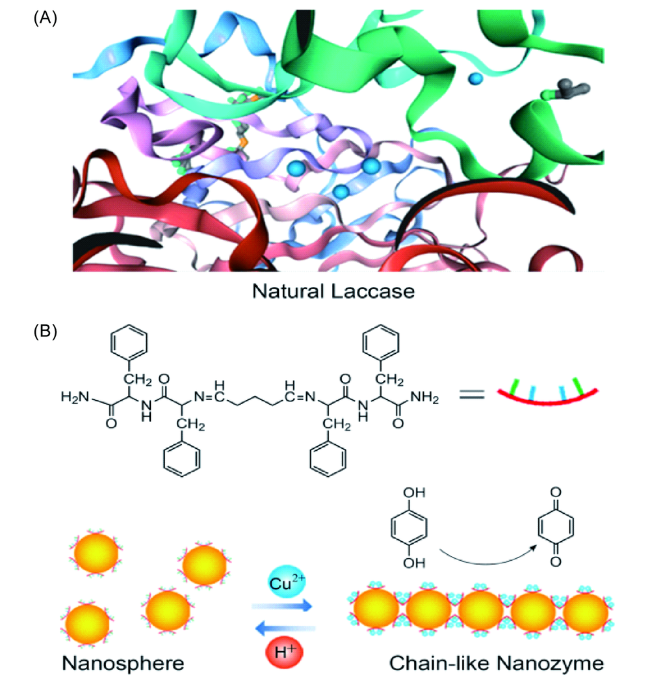
Wang等[33]通过动态共价键组装和金属离子配位作用,获得了链状多级有序结构短肽组装体。通过调节该组装体系的pH能将该纳米链进行解组装,获得稳定的纳米球结构,以此构建了一个结构可逆的短肽基纳米组装体,上述组装体的结构和功能具有离子响应的可逆“开关”性质(图10 )。为了检验组装体的催化活性,将在412 nm处有特定紫外吸收的2-硝基-5-硫代苯甲酸作为指示剂,漆酶可催化氧化对苯二酚为对苯醌,产物与指示剂反应结合后可使指示剂褪色。与天然漆酶相比,这种二肽基纳米组装体作为催化剂在特定时间其催化效率提高了2倍,表现出与底物更高的亲和力,在放置7天后仿生酶仍能保持较高的催化活性,其稳定性远高于天然的漆酶。该研究中的组装体具有了超越天然漆酶的功能,为构筑高催化活性和高稳定性人工酶提供了重要启示[33]。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
图10 (A) 天然漆酶的结构式以及(B) 二肽基超分子装配体在Cu2+和H+的调控下可逆转化及催化过程的示意图[33]Fig.10 (A) Highlighted crystal structure of fungus laccase from Trametes versicolor (PDB: 1GYC). (B) Chemical structure of CDPGA, schematic illustration of reversible transformation of nanospheres to nanochains modulating by Cu2+ and H+, and switchable enzyme-like catalytic oxidation of hydroquinone to benzoquinone[33] |
Chen等[102]描述了一种极简单的方法来构建具有酶活性的基于二肽的纳米超分子结构。生物催化剂是由肽基单元的自组装完成,通过与锌离子的配位,肽自组装过程可以形成超分子β-折叠有序纳米晶体,可以作为基本单元进一步构建更高阶的超分子结构。实验结果显示出了仿生酶具有显著的水解活性和持久的稳定性。该工作进一步说明了使用仿生超分子组装方法开发用于生物技术应用的下一代生物催化剂的可行性和价值。
除了上述应用外,二苯丙氨酸二肽基微纳材料还可以被用于制备各种材料的生物模板、人工光合作用、组织修复材料、喷涂打印、光学制造等方面。
4 结论与展望
FF及其衍生物的组装是一个多因素协同控制的过程,分子间的相互作用和反应发生的外界环境决定了组装体的形貌和结构,通过精准调控可以获得特定形貌和结构的组装体并开展其相关的应用研究。通过向组装体系中引入光敏剂、化药、酶等功能性分子,使肽基微纳材料在生物医药领域有着广泛的应用;向组装体系中引入具有光、电、磁特性的分子,也能进一步拓展其应用范围。
超分子肽共组装是一个新兴的研究领域,可以弥补纯化学合成在一些方面的缺陷,如耗时或者化学合成也难以实现的一些产物。精确设计的多功能肽组装提高了共组装系统内的结构复杂性和化学多样性。动态复杂的共组装途径能够形成具有定制物理、化学和机械特性的组装体,从而拓展了纳米技术的发展空间。此外,这种策略可以有效地扩展微纳材料的应用范围,从设备的制造到疾病的治疗。尽管该研究领域已经取得了重大进展,但肽超分子共组装的全部前景尚未完全实现[103]。比如多个肽基元的有效协同组装以及特定性能调控仍面临着巨大的挑战;在生物医学应用方面,受到生物体系复杂性的影响,理论研究与实际应用之间还存在一定的差距。刺激响应性共组装肽水凝胶和纳米结构同样重要,并且是未来开发先进功能智能纳米材料的方向之一。此外,对具有生物学意义的短肽之间超分子共组装的研究将揭示各种神秘的非共价蛋白质折叠生理学、病理学和药物设计,为人类疾病治疗和健康发展做出贡献。总之,仿生分子材料和系统具有长期潜力,可以带来全新的可持续发展和可降解材料、动态可调传感器、新型自主医疗干预以及新的能源生产和转换模式[104]。随着纳米科学技术、计算设计的进步和自组装、共组装的深入与精细化研究,改进的自动化和人工智能分析技术能够更好地处理复杂系统、异构和动态自适应混合物,基于肽的系统化学最终将产生新的、没有被生物学探索过的解决方案和见解,相信短肽超分子共组装的内在简单性和多方面特性会为纳米科技的发展和应用探索做出更多重要贡献。