




1 研究背景
2 钠离子电池正极材料的掺杂改性
表1 掺杂在钠离子电池正极材料中的应用Table 1 Application of doping in cathode material of sodium ion battery |
| Type | Cathode material | Voltage | Capacity (mAh/g) | Cycle performance | Doping method | ref |
|---|---|---|---|---|---|---|
| Layered metal oxide | NaxMn0.9Co0.1O2 | 1.5~3.8 V | 165(50 mA/g) | 75% (After100 cycle) | Combustion synthesis | 29 |
| NaxFe1/2Mn1/2O2 | 1.5~4.3 V | 190(0.05 C) | 79% (After 30 cycle) | Solid-state reaction | 30 | |
| NaxMn2/3Ni1/3O2 | 2.3~4.5 V | 134(1.7 mA/g ) | 64% (After 10 cycle) | Co-precipitation technique | 31 | |
| Na0.5Mn0.48Co0.5Al0.02O2 | 1.5~4.3 V | 134 (85 mA/g ) | 83% (After 100 cycle) | Sol-gel method | 32 | |
| Na0.9[Cu0.22Fe0.30Mn0.48]O2 | 2.5~4.05 V | 100(0.1 C) | 97% (After 100 cycle) | Solid-state reaction | 33 | |
| NaCr1/3Fe1/3Mn1/3O2 | 1.5~4.2 V | 186(0.05 C) | 54% (After 35 cycle) | Solid-state reaction | 34 | |
| Na0.67Mn0.67Ni0.28Mg0.05O2 | 2.5~4.35 V | 123(0.1 C) | 85% (After 50 cycle) | Sol-gel method | 35 | |
| Prussian blue | NayFe0.4Mn0.1[Fe(CN)6] | 2.0~4.2 V | 119(1 C) | 65% (After 350 cycle) | Ball-milling method | 36 |
| NaxNi0.3Fey[Fe(CN)6] | 2.0~4.0 V | 117(10 mA/g) | 86.3% (After 90 cycle) | Co-precipitation technique | 37 | |
| Na2Mn0.15Co0.15Ni0.1Fe0.6Fe(CN)6 | 2.0~4.0 V | 111(1 C) | 78.7% (After 1500 cycle) | Co-precipitation technique | 38 | |
| Na1.76Ni0.12Mn0.88 [Fe(CN)6]0.98 | 2.0~4.0 V | 118(10 mA/g) | 83.8% (After 800 cycle) | Co-precipitation technique | 39 | |
| Na2Ni0.4Co0.6Fe(CN)6 | 2.0~4.2 V | 92(50 mA/g) | 89.5% (After 100 cycle) | Co-precipitation technique | 40 | |
| Na2CoFe(CN)6 | 2.0~4.1 V | 150(10 mA/g) | 90% (After 200 cycle) | Citrate-assisted controlled crystallization method | 41 | |
| Na0.39Fe0.77Ni0.23 [Fe(CN)6]0.79·3.45H2O | 2.0~4.0 V | 106(10 mA/g) | 96% (After 100 cycle) | Co-precipitation technique | 42 | |
| Polyanionic compounds | NaFePO4@C | 1.5~4.5 V | 145(0.2 C) | 89% (After 6300 cycle) | Electrospinning technique | 43 |
| Br/N/a-C@Na3V2(PO4)3 | 2.5~4.3 V | 83(0.1 C) | 80% (After 500 cycle) | Sol-gel assisted hydrothermal | 44 | |
| Na3Mn1.6Fe0.4P3O11@C | 1.8~4.3 V | 84.9(0.1 C) | 74% (After 100 cycle) | Citric based sol-gel method and carbothermal reduction methods | 45 | |
| Na3V1.9Co0.1(PO4)2F3 | 1.6~4.6 V | 111.3(0.1 C) | 70% (After 80 cycle) | Sol-gel method | 46 | |
| Na3MnTi(PO4)3/C | 1.5~4.2 V | 160(0.2 C) | 92% (After 500 cycle) | Spray-drying method | 47 | |
| Na4MnCr(PO4)3 | 1.4~4.6 V | 160.5(0.05 C) | 74% (After 50 cycle) | Sol-gel method | 48 | |
| Na4Mn3(PO4)2(P2O7) | 1.7~4.5 V | 121(0.05 C) | 86% (After 100 cycle) | Solid-state reaction | 49 |
2.1 掺杂对层状金属氧化物的改性研究
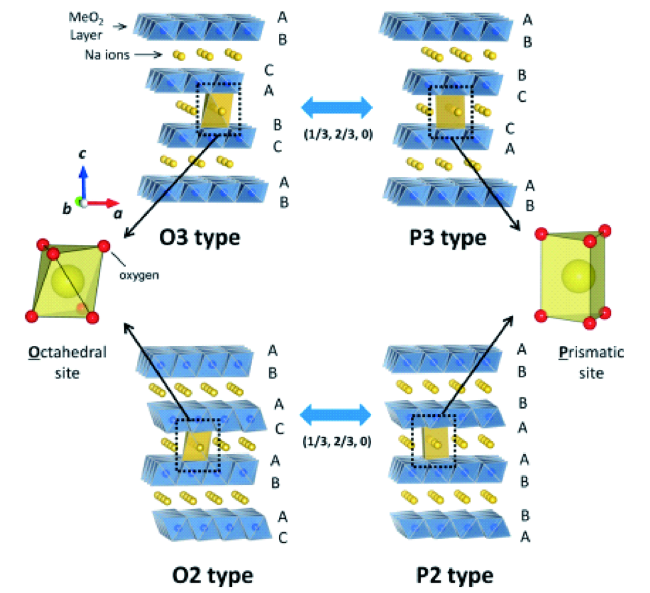
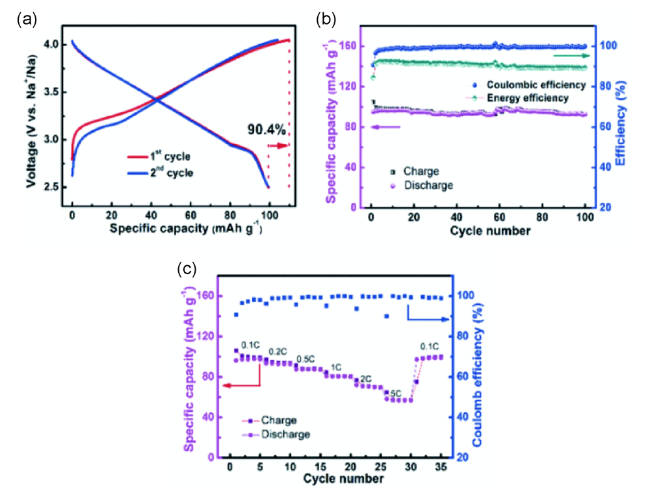
图2 (a) Na0.9[Cu0.22Fe0.30Mn0.48]O2电极的第一和第二恒流充放电曲线在2.5~4.05 V之间以0.1 C (10 mA/g)的速率循环;(b) 在0.1 C速率下的容量、库仑效率和能量转换效率与循环次数的关系;(c) 倍率性能[33]Fig. 2 (a) The first and second constant current charge discharge curves of Na0.9[Cu0.22Fe0.30Mn0.48]O2 electrode were cycled between 2.5~4.05 V at the rate of 0.1 C (10 mA/g); (b) the relationship between the capacity, coulomb efficiency and energy conversion efficiency at the rate of 0.1 C and the number of cycles; (c) the rate performance[33]. Copyright 2015, John Wiley and Sons |
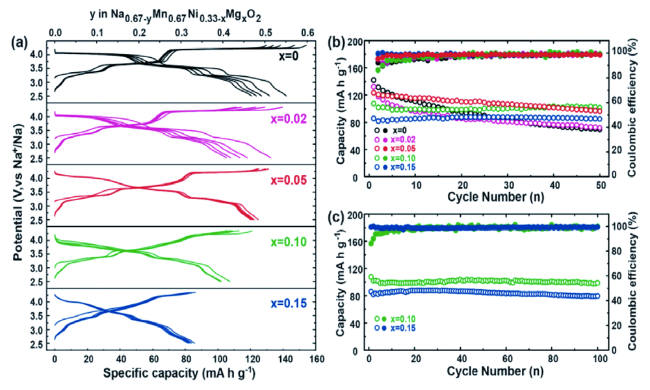
图3 (a) 各种P2型Na0.67Mn0.67Ni0.33-xMgxO2电极(x = 0,0.02,0.05,0.10和0.15)在0.1 C下的恒流充放电电压分布;(b) 各种P2型Na0.67Mn0.67Ni0.33-xMgxO2电极(x = 0,0.02,0.05,0.10,0.15)在50个循环中的循环性能;(c) P2型Na0.67Mn0.67Ni0.33-xMgxO2电极(x = 0.10和0.15)在100圈中的循环性能[35]Fig. 3 (a) Constant current charge discharge voltage distribution of various P2 type Na0.67Mn0.67Ni0.33-xMgxO2 electrodes (x = 0, 0.02, 0.05, 0.10 and 0.15) at 0.1 C;(b) cycling performance of various P2 type Na0.67Mn0.67Ni0.33-xMgxO2 electrodes (x = 0, 0.02, 0.05, 0.10 and 0.15) in 50 cycles;(c) cycling performance of P2 type Na0.67Mn0.67Ni0.33-xMgxO2 electrodes (x = 0.10 and 0.15) in 100 cycles[35]. Copyright 2016, John Wiley and Sons |
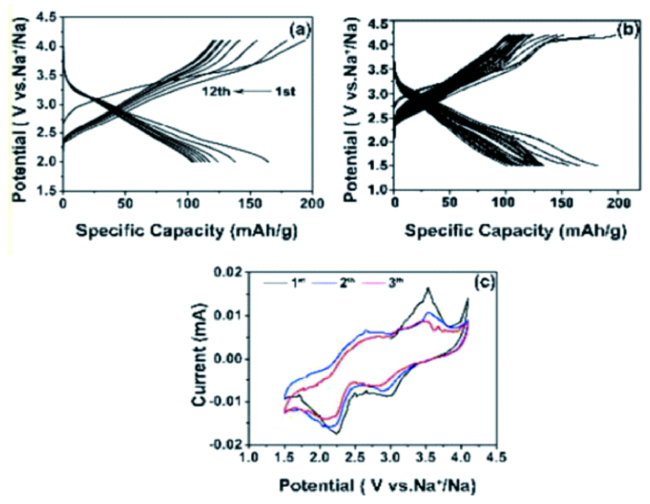
图4 (a) NaCr1/3Fe1/3Mn1/3O2在0.03 C (5 mA/g)电流下的恒流循环曲线与Na+/Na的关系;(b) 在1.5~4.2 V的电位范围内,NaCr1/3Fe1/3Mn1/3O2电极在0.05 C (10 mA/g)下的恒流循环曲线与Na+/Na的关系;(c) NaCr1/3Fe1/3Mn1/3O2电极在1.5~4.1 V之间循环的前三圈循环伏安曲线[34]Fig. 4 (a) The relationship between the constant current cyclic curve and Na+/Na of NaCr1/3Fe1/3Mn1/3O2 electrode at 0.03 C (5 mA/g); (b) the relationship between the constant current cyclic curve and Na+/Na of NaCr1/3Fe1/3Mn1/3O2 electrode at 0.05 C (10 mA/g) in the potential range of 1.5~4.2 V; (c) The first three cycles of cyclic voltammetry of NaCr1/3Fe1/3Mn1/3O2 electrode between 1.5~4.1 V[34]. Copyright 2017, The Royal Society of Chemistry |
2.2 掺杂对普鲁士蓝类材料的改性研究
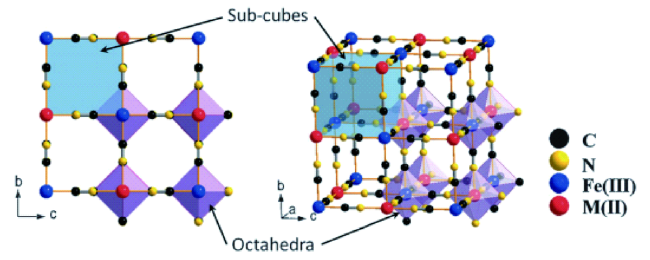
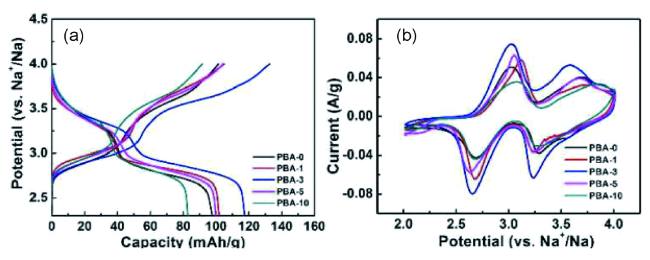
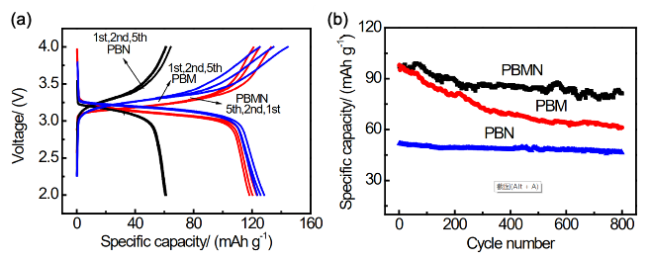
2.3 掺杂对聚阴离子型化合物的改性研究
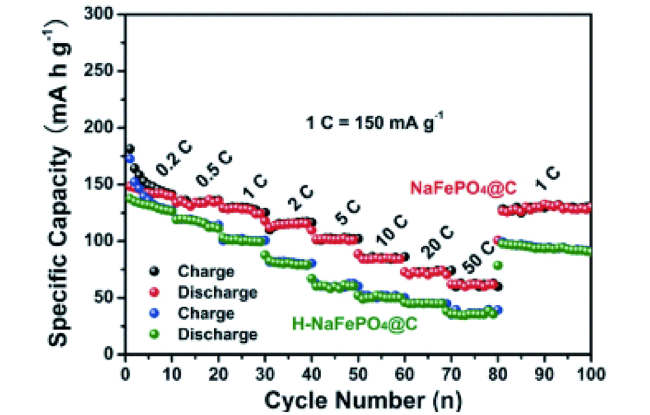
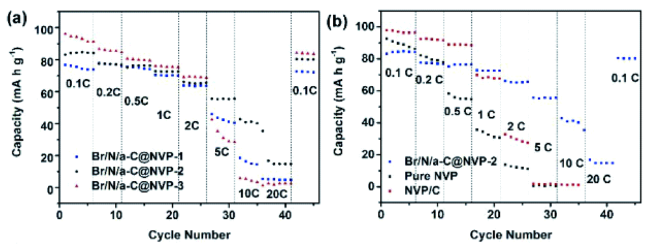
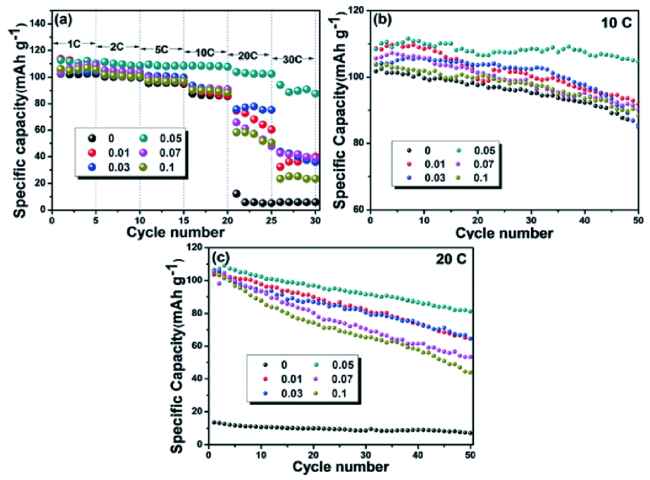
图10 (a) Na3V2-xMgx(PO4)3/C在不同电流密度下的倍率性能; (b) Na3V2-xMgx(PO4)3/C在10 C下的循环性能; (c) Na3V2-xMgx(PO4)3/C在20 C下的循环性能[86]Fig. 10 (a) Rate capability of Na3V2-xMgx(PO4)3/C at different current densities; (b) cycling stability of Na3V2-xMgx(PO4)3/C at 10 C; (c) cycling stability of Na3V2-xMgx(PO4)3/C at 20 C[86]. Copyright 2015, The Royal Society of Chemistry |
3 钠离子电池正极材料的掺杂改性原理
3.1 抑制相变,稳定结构
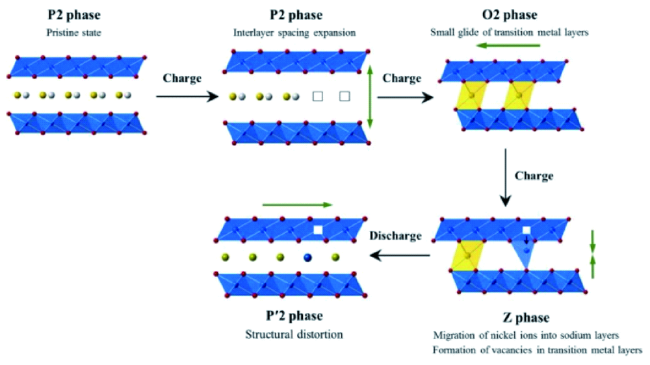
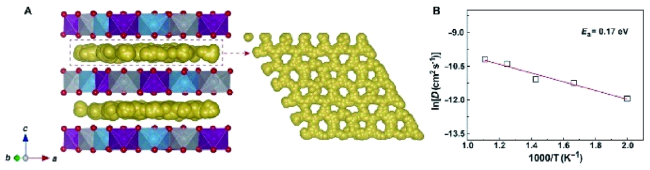
3.2 增大层间距,提升动力学
3.3 提升正极材料的放电容量
3.4 提高材料的电子导电性和离子导电性
表2 三种正极材料的导电性Table 2 Conductivity of three cathode materials |
| Sample | Resistance (Ω·m) | Conductivity(S·m-1) |
|---|---|---|
| Pure ZrO2 | 4.81 × 106 | 2.08 × 10-7 |
| Na0.75Mn0.55Ni0.25Co0.05Fe0.15O2 | 3.21 × 105 | 3.12 × 10-6 |
| Na0.75Mn0.55Ni0.25Co0.05Fe0.10Zr0.05O2 | 1.92 × 105 | 5.20 × 10-6 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}