




1 引言
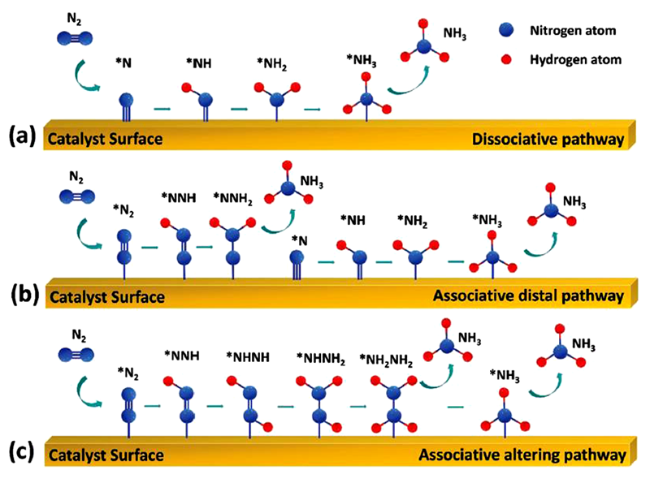
2 NRR的反应机理
3 NRR电催化反应体系的影响因素
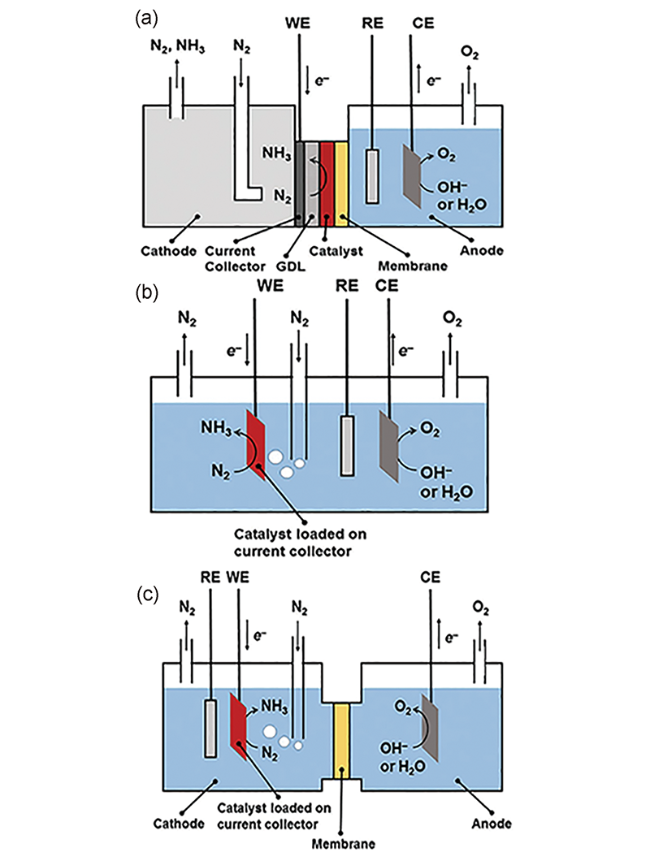
3.1 电解池的设计
3.2 施加电位的影响
3.3 电解质的影响
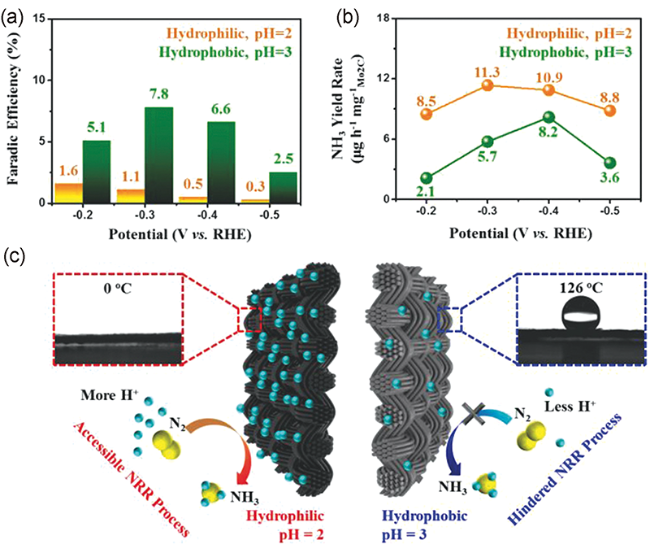
3.3.1 电解液pH值的影响
3.3.2 反离子效应
3.3.3 催化剂与电解质的协同作用
4 NRR的电催化剂研究进展
4.1 贵金属催化剂
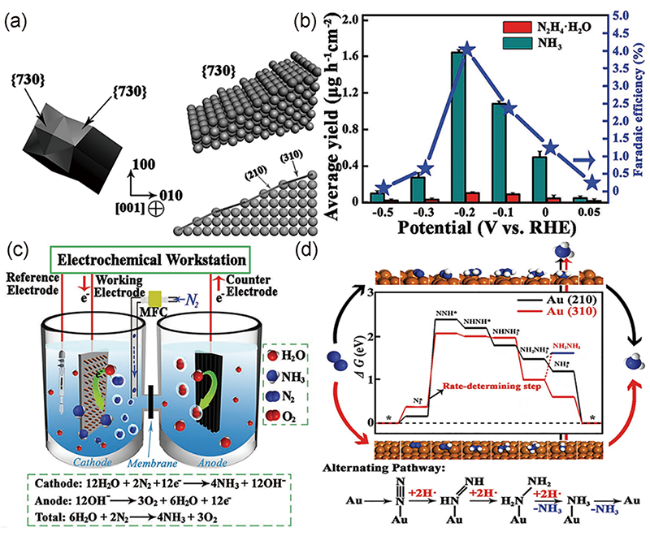
图2 (a) Au THH NRs的TEM图及几何模型;(b) 给定电位下NH3的产率和法拉第效率;(c) NRR的电解池模型;(d) Au THH NRs的自由能图及氮还原催化路径[37]Fig. 2 (a) Transmission electron microscopy image of Au THH NRs and the geometric model. (b) NH3 yield rates and FEs at each given potential. (c) Setup of the electrolytic cell used for the NRR. (d) Free energy diagram and catalytic pathway of N2 reduction on the Au THH NRs[37]. Copyright 2017, Wiley-VCH |
4.2 非贵金属催化剂
4.2.1 过渡金属氧化物
4.2.2 过渡金属氮化物
4.2.3 过渡金属碳化物
4.2.4 过渡金属硫化物
4.3 无金属催化剂
4.4 单原子催化剂
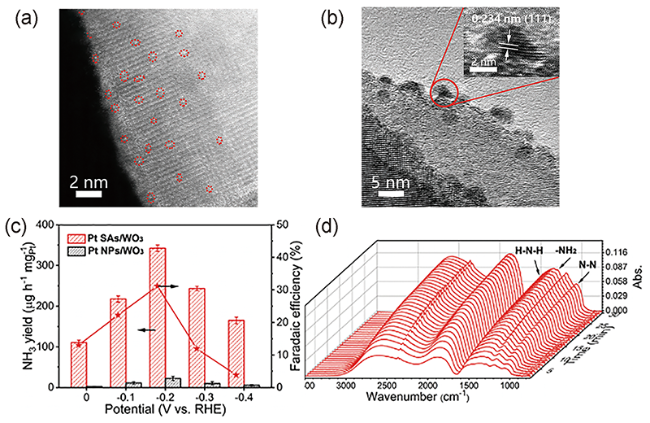
图4 (a) Pt SAs/WO3 的HAADF-STEM图像;(b) Pt NPs/WO3 HRTEM图像;(c) Pt SAs/WO3和Pt NPs/WO3的氨产率和法拉第效率;(d) Pt SAs/WO3的电化学原位时间分辨傅里叶变换红外光谱[94]Fig. 4 (a) HAADF-STEM image of Pt SAs/WO3. (b) HRTEM image of Pt NPs/WO3. (c) Yield of NH3 and Faradaic efficiency of Pt SAs/WO3 and Pt NPs/WO3. (d) Electrochemical in situ time-resolved Fourier transform infrared (FT-IR) spectra for nitrogen reduction reaction (NRR) on the Pt SAs/WO3 electrode[94]. Copyright 2020, Wiley-VCH. |
5 NRR的电催化剂设计
5.1 催化剂的尺寸调控
5.2 催化剂的缺陷工程
5.2.1 空位缺陷
5.2.2 掺杂缺陷

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
图5 (a) Al-Co3O4/NF合成示意图;(b,c) Al-Co3O4/NF的SEM图像;(d) 不同界面结构的NRR模拟过程;(e) Al-Co3O4/NF的电化学原位红外图像;(f) NRR的可能途径[115]Fig. 5 (a) Schematic diagram of Al-Co3O4/NF synthesis. (b, c) SEM images of Al-Co3O4/NF. (d) The proposed limitation and promotion of NRR behavior from different surface interface structures. (e) Electrochemical in situ-FTIR spectra of NRR on Al-Co3O4/NF. (f) Corresponding possible pathway for NH3 production[115]. Copyright 2020, ACS |
5.3 催化剂的形貌调控
5.4 催化剂的非晶化调控
6 结论与展望
表1 NRR电催化剂的总结与对比Table 1 Summary of recently reported NRR electrocatalysts. |
| Catalyst | Electrolyte | NH3 yield rate | FE (%) | Potential (V vs RHE) | ref | |
|---|---|---|---|---|---|---|
| Noble metal- based materials | THH Au nanorods | 0.1 mol/L KOH | 1.648 μg·h -1·c | 4.0 | -0.2 | 37 |
| Hollow Au nocages | 0.5mol/L LiClO4 | 3.9 μg·h -1·c | 30.2 | -0.5 | 120 | |
| porous Au film | 0.1 mol/L Na2SO4 | 9.42 μg·h -1·c | 13.36 | -0.2 | 38 | |
| Au/TiO2 | 0.1mol/L HCl | 21.4 μg·h -1 m | 8.11 | -0.2 | 99 | |
| a-Au/CeOx-rGO | 0.1 mol/L HCl | 8.3 μg·h -1 m | 10.1 | -0.2 | 127 | |
| Pd/C | 0.1mol/L PBS | 4.5 μg·h -1 m | 8.2 | 0.1 | 27 | |
| Pd0.2Cu0.8/rGO | 0.1 mol/L KOH | 2.80 μg·h -1 m | 17.8 | -0.2 | 48 | |
| Pt93Ir7alloy | 0.001 mol/L HCl | 28 μg·h -1·c | 40.8 | -0.3 | 34 | |
| Rh nanosheet | 0.1 mol/L KOH | 23.88μg·h-1·m | 0.217 | -0.2 | 54 | |
| Ag nanosheets | 0.1 mol/L HCl | 4.62×10-11mol ·s -1·cm-2 | 4.8 | -0.6 | 44 | |
| Ag3Cu networks | 0.1 mol/L Na2SO4 | 24.59 μg·h -1 m | 13.28 | -0.5 | 47 | |
| Non-noble metal- based materials | Fe2O3nanorode | 0.1mol/L Na2SO4 | 15.9 μg·h -1 m | 0.94 | -0.8 | 22 |
| Fe/Fe3O4 | 0.1mol/L PBS | 0.19 μg·h -1·c | 8.29 | -0.3 | 57 | |
| MoO3 | 0.1 mol/L HCl | 4.80×10-10 mol·s-1·cm-2 | 1.9 | -0.5 | 59 | |
| Nb2O5 nanofiber | 0.1 mol/L HCl | 43.6 μg·h -1 m | 9.26 | -0.55 | 61 | |
| NbO2 | 0.05 mol/L H2SO4 | 11.6 μg·h -1 m | 32 | -0.65 | 60 | |
| Mn3O4 NP@rGO | 0.1 mol/L Na2SO4 | 17.4 μg·h -1 m | 3.52 | -0.85 | 62 | |
| r-WO3nanosheets | 0.1 mol/L HCl | 17.28 μg·h -1 m | 7 | -0.3 | 63 | |
| WO3-x nanosheets | 0.1 mol/L HCl | 4.2 μg·h -1 m | 6.8 | -0.12 | 64 | |
| Bi4V2O11/CeO2 | 0.1 mol/L HCl | 23.21 μg·h -1 m | 10.16 | -0.2 | 126 | |
| Ti3C2Tx MXene | N.A. | 0.26 μg·h -1·c | 5.78 | -0.2 | 103 | |
| Mo2C/C | 0.5mol/L H2SO4 | 11.3 μg·h -1 m | 7.8 | -0.2 | 79 | |
| MoS2 nanoflower | 0.1mol/L Na2SO4 | 29.28 μg·h -1 m | 8.34 | -0.4 | 107 | |
| Mo2N | 0.1 mol/L HCl | 78.4 μg·h -1 m | 4.5 | -0.3 | 73 | |
| MoN NA/CC | 0.1 mol/L HCl | 3.01×10-10 mol·s-1·cm-2 | 1.15 | -0.3 | 74 | |
| W2N3 nanosheets | 0.1 mol/L KOH | 11.66±0.98 μg·h-1 m | 11.67 ± 0.93 | -0.2 | 76 | |
| Metal-free materials | N-doped carbon | 0.1 mol/L KOH | 57.8 μg·h -1 cm-2 | 10.2 | -0.3 | 118 |
| PCN | 0.1 mol/L HCl | 8.09 μg·h -1 m | 11.59 | -0.2 | 106 | |
| B-doped graphene | 0.05 mol/L H2SO4 | 9.8 μg·h -1·c | 10.8 | -0.5 | 84 | |
| B4C | 0.1 mol/L HCl | 26.57 μg·h -1 m | 15.95 | -0.75 | 88 | |
| Single atom metal materials | Ru SAs/N-C | 0.05 mol/L H2SO4 | 120.9 μg·h -1 m | 29.6 | -0.2 | 92 |
| Ru@ZrO2/NC | 0.1 mol/L HCl | 3665 μg·h -1 m | 21 | -0.21 | 93 | |
| Au1/C3N4 | 0.005 mol/L H2SO4 | 1305 μg·h -1 m | 11.1 | -0.1 V vs Ag/AgCl | 91 | |
| Pt SAs/WO3 | 0.1 mol/L K2SO4 | 342.4 μg·h -1·m | 31.1 | -0.2 | 94 | |
| Mo SAs/N-C | 0.1 mol/L KOH | 34.0±3.6 μg·h-1·m | 14.6±1.6 | -0.3 | 95 | |
| Fe SAs/MoS2 | 0.1 mol/L KCl | 97.5±6 μg·h-1·c | 31.6±2 | -0.2 | 96 |