




1 引言
表1 水系锌离子电池与锂离子电池特征的比较[9,17⇓⇓~20]Table 1 Comparison of characteristics between AZIBs and LIBs[9,17⇓⇓~20] |
| Metal electrode | Ionic radius (Å) | Energy density (W·h·kg-1) | Discharge voltage plateau (V) | Specific capacity (mA·h·g-1) | Volumetric capacity (mA·h·cm-3) | Abundance in earth crust (ppm) | Cost (USD) kg-1 | Safety and recyclability | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lithium | 0.76 | 70~140 | 0.6~1.8 | 3860 | 2061 | 20 | 19.2 | low | ||||||
| Zinc | 0.75 | 180~230 | 3.2~5.0 | 820 | 5855 | 70 | 2.2 | high | ||||||
2 水系锌离子电池锰基正极材料
2.1 MnO2
2.1.1 隧道MnO2
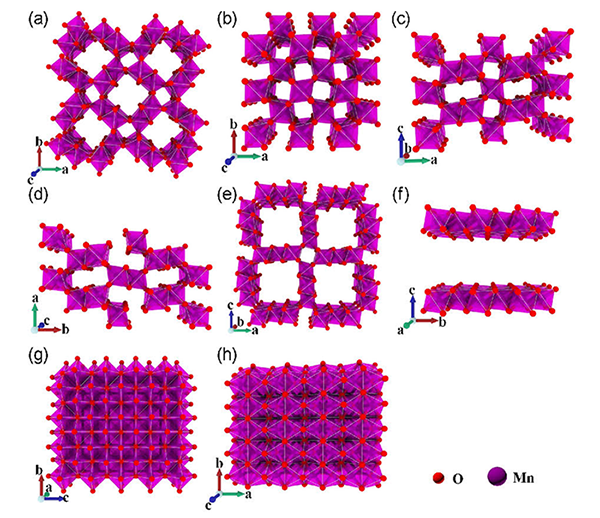
图1 MnO2各种晶型结构示意图: (a) α-MnO 2[31], (b) β-MnO 2[32], (c) γ-MnO 2[33], (d) R-MnO2[9], (e) Todorokite MnO2[34], (f) δ-MnO 2[37] (g) λ-MnO 2[41], (h) ε-MnO 2[32]Fig.1 Schematic diagram of various crystal structures of MnO2: (a) α-MnO 2[31], (b) β-MnO 2[32], (c) γ-MnO 2[33], (d) R-MnO2[9], (e) Todorokite MnO2[34], (f) δ-MnO 2[37] (g) λ-MnO 2[41], (h) ε-MnO 2[32] |
2.1.2 层状MnO2
2.1.3 3D型MnO2
2.2 Mn2O3
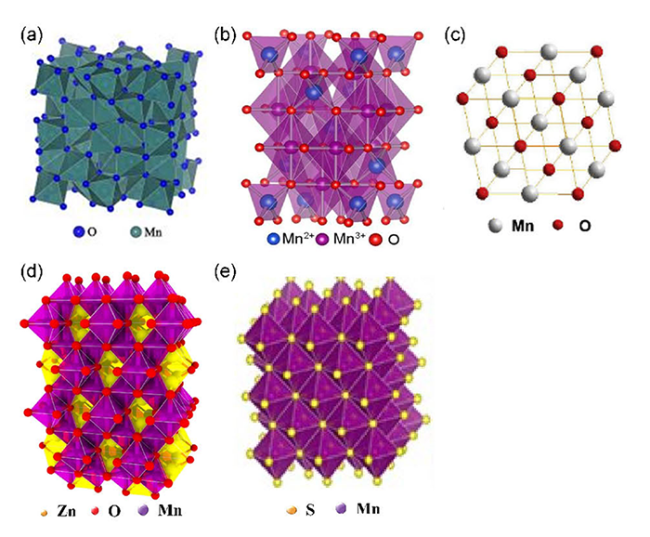
2.3 Mn3O4
2.4 MnO
2.5 其他锰基化合物
2.5.1 ZnMn2O4
2.5.2 MnS
3 锰基水系锌离子电池的储能机理
3.1 Zn2+嵌入/脱出机理
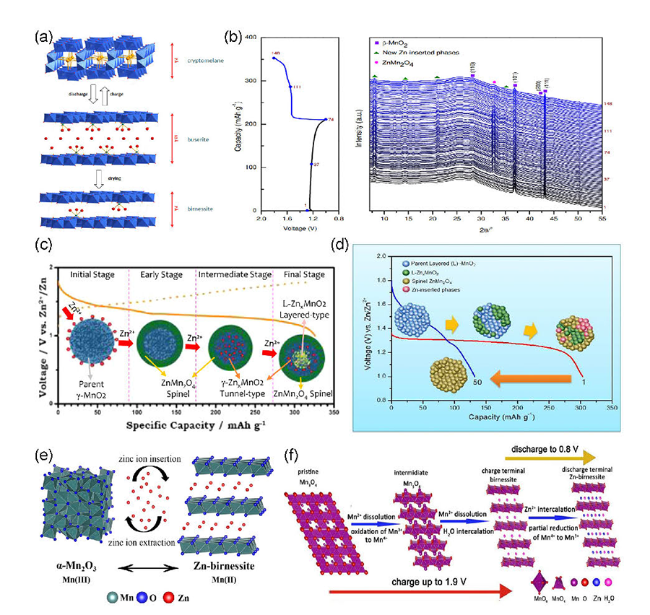
图3 Zn2+在(a) α-MnO 2[36], (c) γ-MnO 2[33], (d) δ-MnO 2[37], (e) α-Mn 2O3[43], (f) Mn3O4[44]中嵌入/脱出的机理示意图[44], (b) β-MnO 2循环过程中的原位XRD图[52]Fig.3 The mechanism of Zn2+insertion/extraction in (a) α-MnO 2[36], (c) γ-MnO 2[33], (d) δ-MnO 2[37], (e) α-Mn 2O3[43], (f) Mn3O4[44], (b) in situ XRD during β-MnO 2 cycle[52] |
3.2 化学转换反应机理
3.3 H+/Zn2+共嵌入/脱出机理
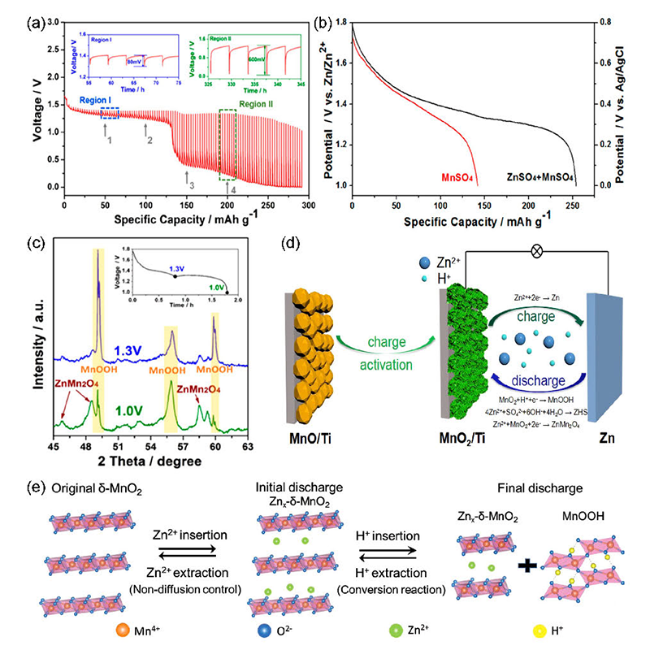
图5 (a) MnO2正极的GITT曲线,(b) 电极在添加ZnSO4和不添加ZnSO4的MnSO4电解液中的放电曲线,(c)放电深度分别为1.3和1.0 V时的XRD图[56],(d) MnO的储能过程示意图[57],(e) δ-MnO 2的储能过程示意图[58]Fig. 5 (a) The GITT curve of MnO2 anode,(b) discharge curve of electrode in MnSO4 electrolyte with or without ZnSO4,(c) the XRD pattern at the discharge depths of 1.3 V and 1.0 V[56],(d) energy storage process of diagram MnO[57],(e) energy storage process of diagram δ-MnO 2[56] |
3.4 溶解/沉积反应机理
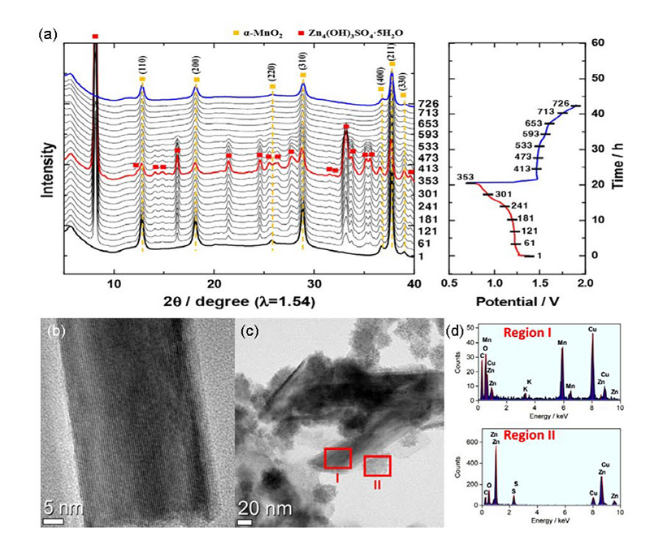

4 锰基正极材料存在的问题和解决策略
4.1 掺杂
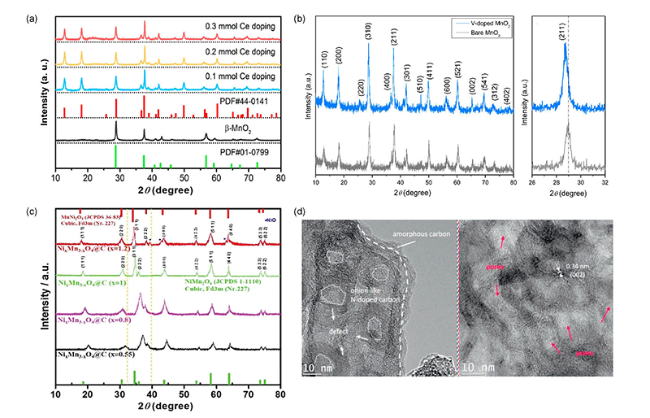
4.2 表面修饰
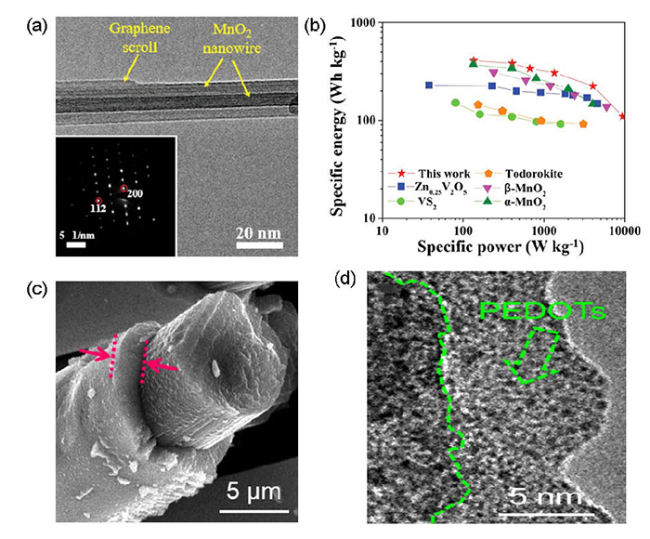
4.3 引入缺陷
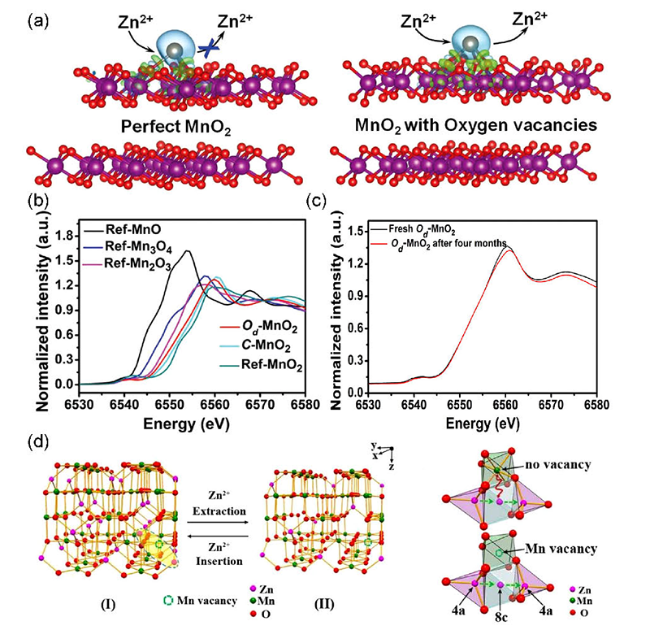
图10 (a) 完整δ-MnO2和Od-MnO2的Zn2+吸附/解吸示意图,(b) Od-MnO2的XANES图,(c) K-边缘X射线吸收近边缘光谱[94],(d) ZnMn2O4尖晶石中嵌入/脱出Zn2+的示意图和ZnMn2O4尖晶石中无锰空位和有锰空位的Zn2+扩散途径[99]Fig.10 (a) The complete Zn2+ adsorption/desorption diagram of δ-MnO 2and Od-MnO2,(b) XANES diagram of od-MnO2,(c) K-edge X-ray absorption near edge spectrum[94],(d) the diagram of Zn2+ embedded/removed from ZnMn2O4 spinel, and the Zn2+ diffusion path without Mn vacancy and with Mn vacancy in ZnMn2O4 spinel[99] |
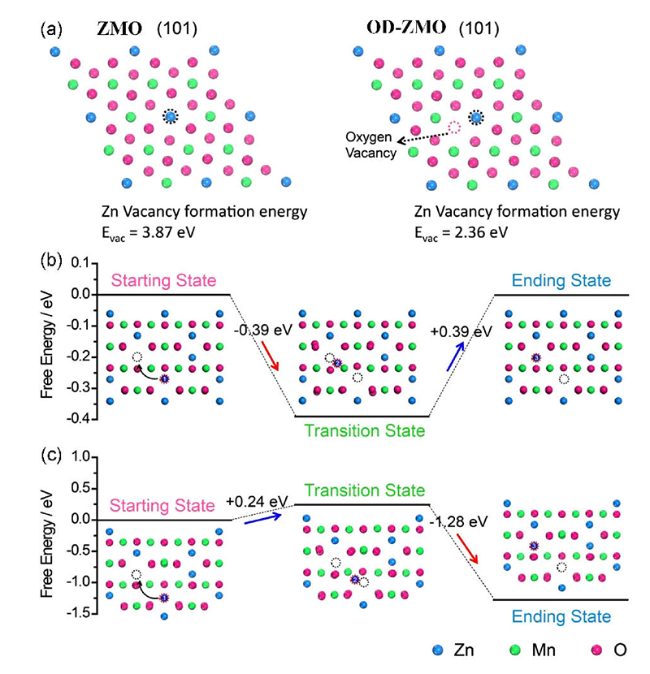
4.4 “支柱”效应
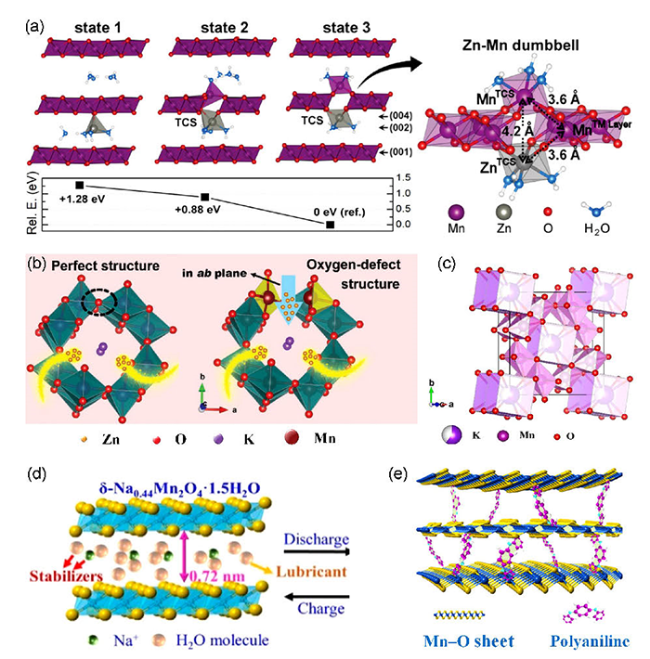
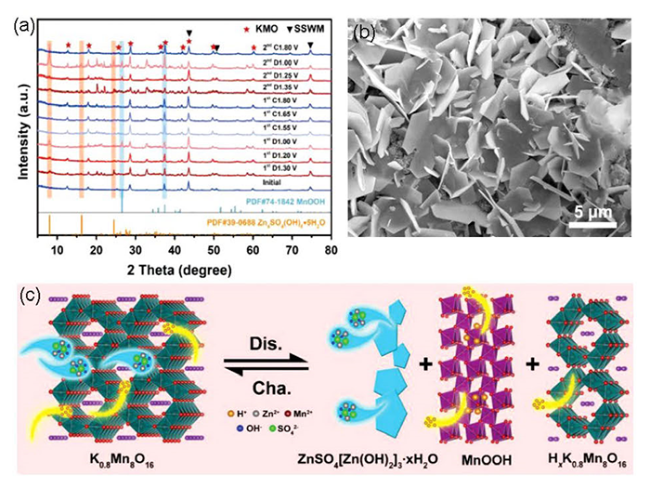
图12 (a) 锌层间结构和相对能量(左边), 形成的锌锰哑铃结构(右边数字是原子间距离)[101],(b) H+扩散到具有完美结构和氧化缺陷的KMO中的示意图[52],(c) KxMn8-xO16结构图[105],(d) Na0.46Mn2O4·1.4H2O结构图[107],(e) PANI嵌入δ-MnO2结构图[111]Fig.12 (a) The structure of zinc interlayer and relative energy(left panel), the formation of Zn-Mn dumbbell structure(right panel number is the distance between atoms)[101],(b) Schematic illustration of H+ diffusion into KMO with perfect structure and oxygen defect structure[52],(c) structure diagram of KxMn8-xO16[105],(d) structure diagram of Na0.46Mn2O4·1.4H2O[107],(e) structure diagram of PANI embedded δ-MnO2[111] |
4.5 复合
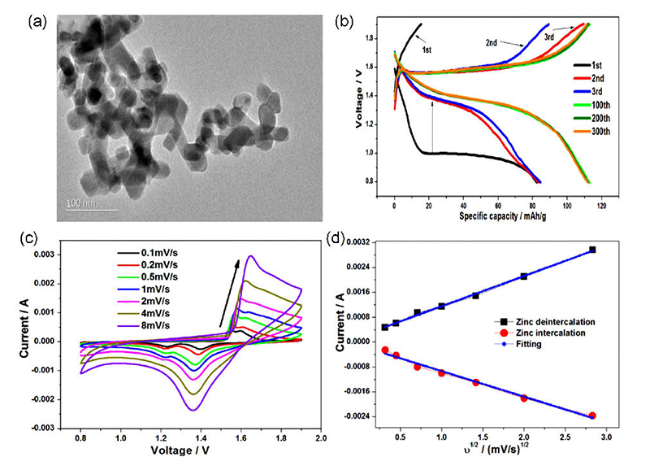
图13 (a) ZnMn2O4/Mn2O3复合材料的TEM图像,(b)恒电流充放电曲线,(c)不同扫描速度下制备的ZnMn2O4/Mn2O3复合材料的循环伏安图,(d)主要正极和负极过程中峰值电流与扫描速度平方根的关系[117]Fig.13 (a) TEM image of ZnMn2O4/Mn2O3 composite,(b) the galvanostatic charge-discharge profiles,(c) the cyclic voltammograms of as-prepared ZnMn2O4/Mn2O3 composite at different scan rates and(d) the relationships between the peak current and square root of scan rate in the main cathodic and anodic processes[117] |
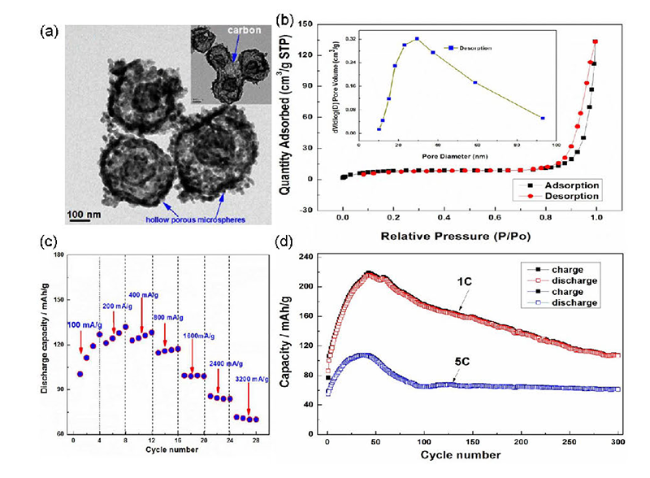
图14 (a) ZnMn2O4的TEM图像,(b)中空多孔ZnMn2O4的N2吸附-解吸等温线和相应的孔径分布曲线(插图),(c)不同电流密度下ZnMn2O4/Zn的倍率性能,(d)不同倍率速率下ZnMn2O4/Zn的循环性能[54]Fig.14 (a) TEM image of ZnMn2O4,(b) N2 adsorption-desorption isotherm of hollow porous ZnMn2O4 and the corresponding pore size distribution curve(inset),(c) The rate performance of ZnMn2O4/Zn at different current densities,(d) The cycling performance of ZnMn2O4/Zn at different C-rates[54] |
4.6 结构设计

图15 SSWM@Mn3O4(a)的合成示意图,(b)的SEM图,(c)的EIS图[122], MnO2@CFP(d)的CFP在工艺上电沉积纳米MnO2的示意图, 以及电沉积前后CFP的相应SEM图,(e)的SEM图, 插图为HESEM图,(f)正极在1.3C下第1、2和100次循环充/放电后的EIS对比图[56]Fig.15 (a) Synthesis schematic,(b) SEM image, and(c) EIS spectra of SSWM@Mn3O4[122], (d) Schematic illustration of the nanocrystalline MnO2 electrodeposited on CFP process, and the corresponding SEM images of CFP before and after electrodeposition,(e) MnO2@CFP of SEM image, inset showing the high-magnified SEM.(f) The EIS comparison diagram of cathode after the first, second, and 100th cycles at 1.3 C[56] |
4.7 电解液优化
4.8 其他策略
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
5 结论与展望
表2 AZIBs化合物中各种锰基材料的结构和电化学性能Table 2 Summary of the configuration and electrochemical performances of various Mn base meterials using in AZIBs |
| Cathode | Morphology | Electrolyte | Voltage(V) | Capacity(mA·h·g-1) | Capacity retention/n cycles/y A·g-1 | ref |
|---|---|---|---|---|---|---|
| ɑ-MnO2 | nanorod | 1 M ZnSO4 | 1~1.8 | 233(83 mA·g-1) | 65%/50 cycles/0.083 | 31 |
| ɑ-MnO2 | nanorod | 2 M ZnSO4 + 0.1 M MnSO4 | 1~1.8 | 161(500 mA·g-1) | 92%/5000 cycles/5 C | 55 |
| ɑ-MnO2@Graphene | nanowire | 2 M ZnSO4 + 0.1 M MnSO4 | 1~1.85 | 382.2(300 mA·g-1) | 94%/3000 cycles/3 | 80 |
| α-MnO2@CNT | nanorod | 2 M ZnSO4 + 0.1 M MnSO4 | 0.8~1.8 | 306(61.6 mA·g-1) | 97%/1000/2.772 | 81 |
| α-MnO2@CNT HMs | microsphere | 2 M ZnSO4 + 0.1 M MnSO4 | 1.2~1.85 | 296(200 mA·g-1) | 97%/1000/2.772 | 138 |
| MnO2@porous-C | nanorod | 2 M ZnSO4 + 0.1 M MnSO4 | 0.8~1.8 | 239(100 mA·g-1) | 100%/1000 cycles/1 | 73 |
| α-MnO2@In2O3 | nanotube | 2 M ZnSO4 + 0.1 M MnSO4 | 1~1.8 | 425(100 mA·g-1) | 75 mA·h·g-1/3000 cycles/3 | 87 |
| Hollow MnO2/CC | nanosheet | 2 M ZnSO4 + 0.1 M MnSO4 | 0.8~1.8 | 263.9(1A·g-1) | 263.9/300 cycles/1 | 123 |
| β-MnO2 | nanorod | 1 M ZnSO4 | 1~1.8 | 270(100 mA·g-1) | 75%/200 cycles/0.2 | 139 |
| β-MnO2 | nanorod | 3 M Zn(CF3SO3)2+ 0.1 M Mn(CF3SO3)2 | 0.8~1.9 | 258(0.65 C) | 94%/2000 cycles/6.5C | 32 |
| β-MnO2@C | nanoparticle | 3 M Zn(CF3SO3)2 + 0.1 M MnSO4 | 1.0~1.8 | 150(200 mA·g-1) | 100%/400 cycles/0.3 | 74 |
| γ-MnO2 | mesoporous | 1 M ZnSO4 | 0.8~1.8 | 285(0.05 mA·c ) | 63%/40 cycles/0.5mA·c | 33 |
| γ-MnO2@C | nanorod | 2 M ZnSO4 + 0.4 M MnSO4 | 0.8~1.8 | 301(500 mA·g-1) | 64.1%/300 cycles/10 | 75 |
| Todorokite-MnO2 | nanoflake | 1 M ZnSO4 | 0.7~2.0 | 108(0.5 C) | 72%50 cycles/0.5C | 34 |
| δ-MnO2 | nanoflake | 1 M ZnSO4 | 1.0~1.8 | 252(83 mA·g-1) | 44%/100 cycles/0.1 | 38 |
| δ-MnO2 | nanoparticle | 1 M Zn(TFSI)2+0.1 M Mn(TFSI)2 | 0.9~1.8 | 238(0.2 C) | 93%/4000 cycles/20C | 58 |
| λ-MnO2 | nanoparticle | 1 M ZnSO4 | 1~1.8 | 136(100 mA·g-1) | - | 41 |
| ε-MnO2@CFP | nanoparticle | 2 M ZnSO4 + 0.2 M MnSO4 | 1~1.8 | 290(1 C) | 100%/10 000 cycles/6.5C | 56 |
| ɑ-Mn2O3 | nanoparticle | 2 M ZnSO4 + 0.1 M MnSO4 | 1~1.9 | 148(100 mA·g-1) | 51%/2000 cycles/0.1 | 43 |
| Mn2O3/Al2O3 | microbundle | 2 M ZnSO4 + 0.1 M MnSO4 | 1~1.8 | 289(300 mA·g-1) | 118 mAh·g-1/1100 cycle/1.5 | 117 |
| Mn3O4 | nanoparticle | 2 M ZnSO4 | 0.8~1.9 | 239.2(1 A·g-1) | 73%/300 cycles/0.5 | 44 |
| Porous cube-like Mn3O4 @C | porous nanocube | 2 M ZnSO4 + 0.1 M MnSO4 | 0.8~1.9 | 102.3(2 A·g-1) | 77.1%/200 cycles/0.5 | 119 |
| Mn3O4@NC | nanorod | 2 M ZnSO4 + 0.1 M MnSO4 | 0.8~1.9 | 280(100 mA·g-1) | 77.6%/700 cycles/1 | 77 |
| MnO | nanoparticle | 2 M ZnSO4 + 0.1 M MnSO4 | 1.0~1.9 | 330(100 mA·g-1) | 300 mAh·g-1/300 cycles/0.3 | 57 |
| Mn0.61□0.39O @C | nanoparticle | 2 M ZnSO4 + 0.1 M MnSO4 | 0.8~1.8 | 117.2(1 A·g-1) | 99%/1500 cycles/1 | 45 |
| ZnMn2O4 | microrod | 1 M ZnSO4 + 0.1 M MnSO4 | 0.6~1.9 | 240(200 mA·g-1) | 79%/1000 cycles/2 | 61 |
| Hollow ZnMn2O4 | hollow microsphere | 2 M ZnSO4 + 0.1 M MnSO4 | 0.8~1.9 | 220(100 mA·g-1) | 106.5 mAh·g-1/300 cycles/0.1 | 54 |
| ZnMn2O4/C | nanoparticle | 3 M Zn(CF3SO3)2 | 0.8~1.9 | 150(50 mA·g-1) | 94%/500 cycles/0.5 | 99 |
| ZnMn2O4@PCPs | nanoparticle | 1 M ZnSO4 +0.05 M MnSO4 | 0.8~1.8 | 145.2(1000 mA·g-1) | 86.5%/2000 cycles/1 | 121 |
| HM-ZnMn2O4@rGO | hollow nanosphere | 2 M ZnSO4 + 0.1 M MnSO4 | 0.8~1.8 | 147(300 mA·g-1) | 72.7/6500 cycles/1 | 140 |
| OD-ZnMn2O4@PEDOT | nanofiber | PVA/LiCl/ZnCl2/MnSO4Gel | 0.8~1.9 | 221(0.5 mA·c ) | 93.8%/300 cycles/8 mA·c | 84 |
| MnS | nanoparticle | 2 M ZnSO4 | 1~1.8 | 110(500 mA·g-1) | 63.6%/100 cycles/0.5 | 50 |
| MnOx@N-C | nanorod | 2 M ZnSO4 + 0.1 M MnSO4 | 0.8~1.8 | 100(2 A·g-1) | 100 mA·h·g-1/1600 cycles/2 | 53 |
| V-doped MnO2 | nanoparticle | 1 M ZnSO4 | 1~1.8 | 266(66 mA·g-1) | 49%/100 cycles/0.066 | 70 |
| Ce-doped α-MnO 2 | nanorod | 2 M ZnSO4 + 0.1 M MnSO4 | 1~1.8 | 134(5 C) | 70%/100 cycles/5 C | 68 |
| Ti-MnO2 | nanowire | 3 M Zn(CF3SO3)2+ 0.1 M Mn(CF3SO3)2 | ~ | 259(100 mA·g-1) | 86%/4000 cycles/1 | 71 |
| NixMn3-xO4@C | nanoparticle | 2M ZnSO4 + 0.15 M MnSO4 | 1~1.85 | 133.7(200 mA·g-1) | 91.9%/850 cycles/0.4 | 72 |
| ZnNixCoyMn2-x-yO4@N-GO | nanoparticle | 2 M ZnSO4 +0.1 M MnSO4 | 0.7~1.7 | 3.5(1.5 A·g-1) | 79%/900 cycles/1 | 121 |
| H2O-intercalated δ-MnO 2 | nanoflake | 1 M ZnSO4 | 1~1.9 | 154(3 A·g-1) | 75.3%/200 cycles | 101 |
| Na0.46Mn2O4·1.4H2O | nanoplates | 2 M ZnSO4 + 0.2 M MnSO4 | 0.9~1.9 | 159(2 A·g-1) | 98%/10 000 cycles/20 C | 107 |
| La+-intercalated δ-MnO 2 | nanofloret | 1 M ZnSO4 + 0.4 M MnSO4 | 0.8~1.9 | 278.5(100 mA·g-1) | - | 108 |
| K0.8Mn8O16 | nanorod | 2 M ZnSO4 + 0.2 M MnSO4 | 1~1.8 | 330(100 mA·g-1) | 150 mAh·g-1/1000 cycles/1 | 52 |
| KxMn8-xO16 | nanodendrite | 1 M Zn(CF3SO3)2 + 0.05 M MnSO4 | 0.8~1.8 | 116(100 mA·g-1) | 74%/300 cycles/0.1 | 105 |
| P-MnO2-x@VMG | nanosheet | PVA/ZnCl2/MnSO4 Gel | 1~1.8 | 302.8(500 mA·g-1) | 90%/1000 cycles/2 | 109 |
| ZnMn2O4/NG | nanoparticle | 1 M ZnSO4 +0.05 M MnSO4 | 0.8~1.8 | 221(100 mA·g-1) | 97.4%/2500 cycles/1 | 76 |
| MnOx/PPy | nanoparticle | 2 M ZnSO4 + 0.1 M MnSO4 | 0.4~1.9 | 302(150 mA·g-1) | 114 mAh·g-1/1000 cycles/6 | 141 |
| ZnMn2O4/Mn2O3 | microsphere | 1 M ZnSO4 | 0.8~1.9 | 82.6(500 mA·g-1) | 112 mAh·g-1/300 cycles/0.5 | 117 |
| Binder-free Mn3O4 | nanoflower | 2 M ZnSO4 + 0.1 M MnSO4 | 1~1.8 | 296(100 mA·g-1) | 100%/100 cycles/0.5 | 122 |
| oxygen-deficient δ-MnO 2 | nanosheet | 1 M ZnSO4 + 0.2 M MnSO4 | 1~1.8 | 159(200 mA·g-1) | 80%/3000 cycles/5 | 94 |
| oxygen-deficient β-MnO 2 | nanowire | 2 M ZnSO4 + 0.1 M MnSO4 | 0.8~1.8 | 302(50 mA·g-1) | 94%/3000 cycles/5 | 95 |