



1 生物大分子聚集的生理及病理现象
生物大分子的凝聚态及其动态变化过程,涉及许多重要的生理及病理过程。如凝血现象,分别涉及一系列凝血因子的活化、凝血斑块形成、血栓溶解等生理动态过程。阿尔茨海默病患者脑内β淀粉样蛋白(amyloid-β,Aβ)沉积则是其标志性的神经病理学特征之一。
1.1 凝血现象
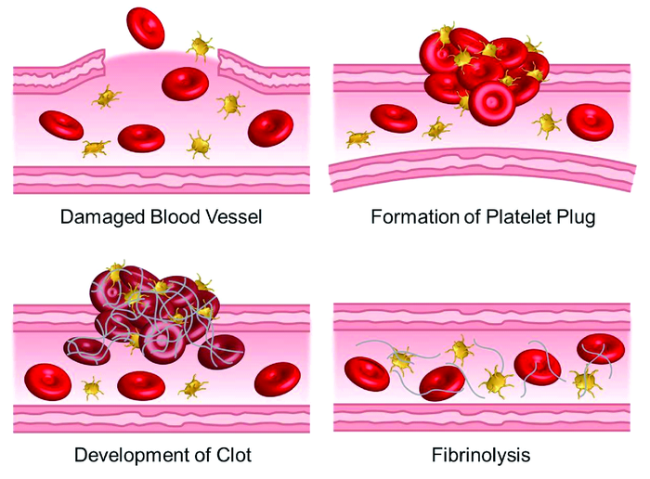
凝血斑块的形成涉及一系列重要的细胞和生物化学分子事件[1, 2]。凝血瀑布模型约在1960年首次提出,模型中指出凝血过程能够被两种通路(外源性和内源性通路)分别激活。启动外源性通路需要血浆暴露于组织因子(TF,也称促凝血酶原、凝血因子Ⅲ或CD142)中,组织因子通常存在于血管外组织,即在血液外部。而内源性的激活方式则依赖于多种存在于血液内部的因子;在血浆与许多人工表面(如高岭土、微粉硅等)接触时也会激活内源性通路。一旦凝血过程被上述任意一个通路激活,单一凝血因子便可以在凝血级联或“瀑布”被顺序的放大激活。凝血酶(Thrombin)又称凝血因子IIa,是机体凝血系统的一种关键酶,是凝血瀑布级联中的核心。它一方面可直接作用于血液中的纤维蛋白原,使之转变为纤维蛋白,形成纤维蛋白凝胶,另一方面则通过受体介导途径诱导血小板活化,活化的血小板发生聚集,在纤维蛋白凝胶网的作用下形成血小板血栓,之后血液中其他有形成分的加入导致稳定血栓的形成。因此,凝血酶在不需要其他众多凝血因子参与的情况下,可快速而高效的诱导血栓的形成,是一种有效的促凝剂[1,2,3]。
1.2 阿尔茨海默病
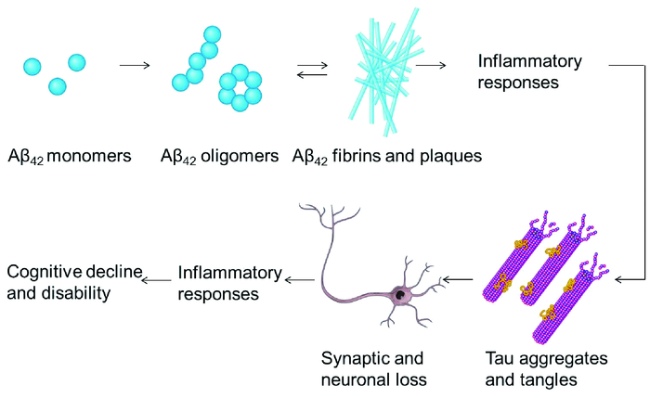
研究表明阿尔茨海默病患者脑部均有一些显著的神经病理学特征,例如β淀粉样蛋白(Amyloid-β,Aβ)沉积、异常磷酸化tau蛋白导致的神经纤维缠结、基底前脑胆碱能神经元变性以及基因突变等。而这其中Aβ自发产生聚集,进而形成有毒的低聚物与纤维,被认为是导致AD患者突触损伤和记忆缺陷的主要因素[5, 7]。Aβ是由淀粉样前体蛋白(Amyloid precursor protein,APP),即一种695~770个氨基酸的I型跨膜糖蛋白代谢产生的。Aβ存在各种长度,40个氨基酸长度构成的多肽片段Aβ40和溶解度较低的42个氨基酸长度多肽Aβ42最为丰富。Aβ42与Aβ40相比,在羧基端多出两个氨基酸(Ile41,Ala42),表现出了更强的成纤维能力。脑内Aβ肽聚集被认为是阿尔茨海默病的最初事件,发生于出现临床症状的15~20年之前,主要由于脑内清除这些多肽的功能缺陷而产生。淀粉样蛋白假说是关于阿尔茨海默病致病机理的一种重要假说[5, 7]。这种假说认为,Aβ42蛋白的增加会触发大脑中导致阿尔茨海默病的一系列事件。Aβ42单体聚集形成寡聚集体、纤维及Aβ42斑块(AD的病理学标志之一)。Aβ聚集起始于海马(Hippocampus)和内嗅皮层(Entorhinal cortex),而这些Aβ的聚集导致了一系列的炎症反应。虽然具体过程仍尚未定论,但这些事件仍导致了tau蛋白(一种微管相关蛋白)的聚集和异常磷酸化、神经纤维缠结(Neurofibrillary tangles,NFTs)。此外,神经原纤维缠结中磷酸化tau蛋白的细胞内沉积还导致进行性细胞骨架变化,破坏轴突运输。由此受损的神经元和突触则功能失调、死亡,进而导致炎症反应,神经元和突触进一步的功能损伤和退行性病变导致病人认知功能下降与逐渐失能(如图2所示)。
目前对于AD的治疗策略是姑息性的,如使用乙酰胆碱酯酶抑制剂(Acetylcholinesterase inhibitors,AChEIs)抑制胆碱(Acetylcholine,Ach)降解,维持神经传导水平,且仅在病人认知能力和运动功能继续退化之前的短期内控制症状。然而,这种治疗方法最终还是会失效,病人最终认知功能还是逐渐下降,导致失能[8]。目前,发展有力的治疗手段仍然面临着极大的挑战,对疾病产生机制的透彻认识将有助于发展针对性的治疗策略。目前对于AD治疗的另一问题是,病理学事件在疾病确诊之前就已经开始,在此时进行干预可能会阻止进一步的退行性变化,因此发展准确的早期检测方法至关重要。同时,AD是脑部疾病,还需解决跨血脑屏障(Blood-brain barrier,BBB)的药物递送问题[9]。
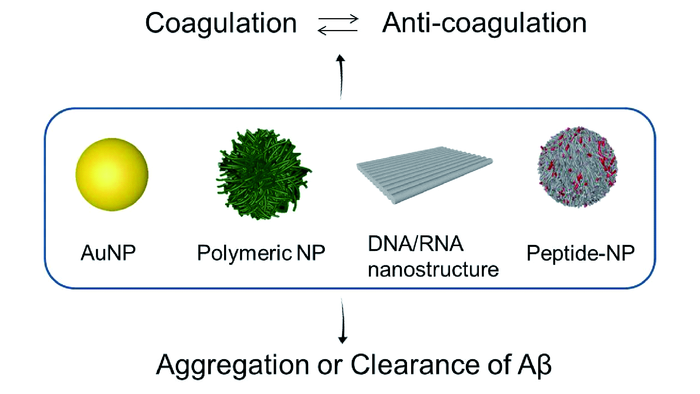
2 对生物凝聚态具有调控作用的纳米材料
基于原子或分子组装,纳米技术可构建出新的纳米功能器件或具有新生物效应的纳米材料,并在纳米尺度上对物理、化学、材料和生命科学等提供新的研究视角和技术手段[10,11,12,13]。近年来,纳米材料和医学技术快速发展,为解决生物医学领域的重大问题提供了强有力的研究手段,并逐渐形成了纳米生物医学的研究领域。经过多年实践,实验室和临床研究中不断涌现大量功能多样并具有应用潜力的纳米材料[10,11,12,13]。许多纳米材料可以用于制备纳米药物或纳米药物运输载体,为疾病诊断、治疗、调控的有效成分,将其有效递送至病灶部位并合理释放发挥功能,提供了有力的工具。目前已有多种含有纳米材料(或纳米成分)的药物制剂和载体系统进入临床试验阶段,也有部分纳米药物已经用于临床的诊断与治疗。
利用纳米技术调控生物大分子凝聚态,发展新颖的疾病诊断和治疗策略,具有非常显著的特点与优势:(1)首先,纳米材料物理、化学及生物特性是其可以直接用于调控生物凝聚态、进行疾病诊断治疗的重要保证。利用纳米技术,能够有效控制多种多样纳米材料的尺度、形状和表面修饰等关键理化特性,能够更好的利用其装载运输药物,如改善药物水溶性、提高其稳定性并发挥药效。(2)多种多样可控制备的纳米材料除了可以有效的装载传统小分子药物,还能够装载多肽、蛋白、抗体、酶类药物和功能核酸等多种不同的功能成分,用于生物凝聚态调控及疾病的诊断与治疗。(3)多种多样可控制备的纳米材料通过设计实现高效、安全、靶向的药物运输、缓慢或响应性控制的药物释放,能够更有效的作用于多种生物大分子,实现有效的凝聚态调控和疾病诊断与治疗。(4)多靶点药物联用是当今包括肿瘤在内多种疾病治疗中非常有前途的发展方向。针对具体的生物凝聚态过程,如凝血斑块形成或淀粉样蛋白聚集现象,联合使用多种调控作用成分,能够多靶点协同增强调控或治疗效果,并减少毒副作用。多功能纳米材料还可同时装载调控成分与影像成分,实现在调控凝聚态的同时对疾病的检测与疗效进行监控,实现诊疗一体化的最佳效果。
利用纳米技术制备多种纳米颗粒或制剂,在调控生物大分子的凝聚态过程、进行疾病的诊断与治疗方面具有多种优势,利用纳米材料特性进行合理设计和优化,有望改善药物水溶性、提高药物稳定性、实现药物缓慢控制释放、病灶靶向定位递送、多靶点治疗和诊疗一体化等。总之,纳米技术为实现生物凝聚态调控、实现疾病的诊断与治疗提供了新的机遇。
2.1 金属纳米颗粒
金属纳米颗粒是用于药物载体设计的常见纳米颗粒之一[12, 14]。由惰性金属(金或钛)构成的纳米颗粒已广泛用于临床前阶段研究,如药物的装载、运输以及可控释放中。金属纳米颗粒具有各种独特的性质,也在许多方面展示了优势。金纳米颗粒是其中的一大类,使用最为广泛的是球形和棒状的金纳米颗粒[14]。金纳米颗粒最为独特的性能是其局域等离子体共振现象(Localized surface plasmon resonance,LSPR),即金颗粒表面价电子与特定频率入射光产生震荡效应,在颗粒的近场区域产生强烈的电磁场,在光谱上出现一个强的共振吸收峰,可以用于化学传感器和生物传感器的构建。球形金纳米颗粒的吸收峰主要在520 nm,用于活体水平时,该波长处光会被生物组织迅速衰减。棒状金纳米颗粒LSPR具有两个长轴和短轴两个方向。通过调整金纳米棒的长径比,其最大吸收峰能够从可见光区移动到近红外区(650~1300 nm),适用于活体水平研究。金纳米颗粒可以用作药物运输的载体、多种模式的成像造影剂及深层组织的光热治疗等[14]。虽然这些金属纳米颗粒都具有惰性与一定程度的生物相容性,但其在活体水平的滞留效应仍不可忽视,反复给药后金属纳米颗粒的积累可能会导致毒副作用。目前大部分利用金属纳米颗粒进行药物递送的工作仍在临床前阶段[12]。
2.2 高分子纳米颗粒
高分子纳米颗粒多由生物相容性较好、能够生物降解的高分子材料构建而成,如聚乙二醇和聚乙烯吡咯烷酮等。大部分高分子纳米颗粒利用具有两段或多段不同疏水性聚合物链的嵌段共聚物自组装形成[12, 15]。这些共聚物在水溶液中会自组装形成具有核壳结构的纳米颗粒,其中疏水嵌段形成核心,以减小其在水溶液环境中的暴露,而亲水部分形成外壳起稳定核心的作用。这种核壳结构的纳米颗粒能够将药物装载在疏水核心,可以达到很高的药物装载效率,亲水壳层可以提供很好的空间保护作用,很适合于装载药物并进行运输。高分子纳米材料目前已被用于包载亲水或疏水性药物小分子、生物大分子如蛋白质或核酸等。除自组装技术外,高分子纳米颗粒也可以通过使用带有微小模具的薄膜,利用非润湿模板的粒子复制(Particle replication in nonwetting templates,PRINT)技术制备,从而获得一系列具有精确尺寸、形状和化学组成的高分子纳米颗粒。环境响应型高分子或智能高分子材料,是由多种线形和支化(共)聚合物或交联聚合物网络组成的一类材料。这类响应型高分子材料具有对外界刺激产生物理化学行为改变的能力,如温度和pH变化常被用作响应型高分子纳米颗粒的刺激因素,除此之外,常见的刺激因素还包括超声、离子强度、氧化还原状态、电磁信号辐射和生物化学信号等;而高分子材料理化行为改变的类型包括溶解度、亲水性-疏水性平衡和构象的转变,能够用于响应型的药物释放。许多高分子纳米材料在临床研究以及临床前实验研究中已经广泛开展[12, 15]。
2.3 自组装核酸纳米材料
核酸纳米技术以DNA/RNA大分子作为自组装纳米材料的基元,由于分子间相互作用特性,通过化学键自发组合,可形成结构稳定、复杂有序并且具有某种特定功能的超分子结构或分子聚集体[16]。其中DNA/RNA折纸术(DNA/RNA origami)是一种新颖而独特的自组装核酸纳米策略,可用来组装完全可识别的和大小可控的纳米结构[17, 18]。DNA折纸技术是将大约7000个碱基的单链DNA(骨架链,Scaffold strand)与一系列经过序列设计的不同的DNA短链(辅助折叠链,Helper strands)通过碱基互补折叠,可控的构造出所设计的纳米图形结构,现在已被广泛用来制作各种纳米尺度的二维DNA图案、形状和三维复杂结构。由于构成DNA折纸的所有辅助折叠链都是不同的,其位置完全取决于所设计的序列,从而使整个DNA纳米阵列在空间上完全可寻址[17]。在核酸纳米结构构建时,可根据需要预先设计其纳米形状和尺寸,得到满足功能需求的纳米结构;又可以针对核酸纳米结构组成链或其互补链进行修饰,使之能够与功能材料基团有效的连接,最终在纳米结构表面或内部构建形成预先设计的、可控的多种功能材料的自组装体,具有装载包括小分子染料、基因片段、金属纳米粒子和蛋白质等“货物”的能力。由于自组装核酸纳米材料具有结构精确可控、易于修饰且生物可降解的特点,引起生物纳米材料领域的广泛关注[19, 20]。
2.4 自组装多肽纳米材料
天然蛋白质中,多肽链的氨基酸排列顺序即为蛋白质的一级结构,通过这些氨基酸序列之间非线性的相互作用形成β片层,α螺旋或无规卷曲结构,形成蛋白质的二级结构。这些二级结构基序可以通过相互作用形成复杂的高级结构。经过合理设计,可通过利用β片层,α螺旋构建模块化、可调节的自组装多肽纳米结构[21]。根据多肽片段的不同,这些基于多肽的纳米结构也可以经过偶联、延伸和修饰后具有不同的生物功能。如β片层形成肽Q11(QQKFQFQFEQQ)可自组装形成纳米纤维,已被用于纳米疫苗的制备,可通过固相合成在多肽抗原的C末端连接Q11多肽,Q11自组装成β片层进而形成纳米纤维时,抗原多肽则可被连接于纤维的外表面[22]。α螺旋也可作为一种基序用于构建纳米纤维或纳米颗粒。如使用具有α螺旋Coil29,利用固相合成技术将抗原表位与之偶联,当Coil29组装成纳米纤维后,抗原表位可以垂直于纤维分布。通过预先设计,将α螺旋五聚体和三聚体形成多肽片段偶联起来,经过重组表达后得到的多条长多肽链段经自组装后能够形成纳米颗粒[23]。多肽也可以与磷脂分子或其他高分子共价偶联,通过自组装形成多肽两亲性胶束(Peptide amphiphile micelles,PAMs)等纳米结构[21]。基于多肽序列的可设计性、可控的自组装性能以及良好的功能化潜力,自组装多肽纳米材料在纳米药物载体设计和递送、疫苗设计及免疫治疗方面具有良好的应用前景。
3 纳米材料对生物凝聚态的调控
纳米材料经过合理的功能化修饰后,具备对生物凝聚态调控的能力。一些研究工作报道显示,纳米材料能够作为血液的促凝剂、抗凝剂使用,或利用其对于淀粉样蛋白聚集状态的识别、结合、调控能力而作为阿尔茨海默病早期检测与治疗手段使用。以下将以纳米材料对凝血现象的调控和对阿尔茨海默病的诊断与治疗为例进行讨论。
3.1 纳米材料对凝血现象的调控——促进血液凝聚
肝动脉灌注化疗栓塞(Transcatheter arterial chemoembolization,TACE)是目前公认的肝癌非手术疗法的首选方法,利用导管选择性的插入到肿瘤动脉血管中,注入栓塞剂阻塞血管或减缓血流,引起肿瘤组织缺血坏死,如果同时使用化疗药物与微球联合,则起到化疗栓塞作用,是目前肝癌患者最主要的治疗手段之一[26, 27]。这种导管靶向的肿瘤血管栓塞疗法虽然使用广泛,但仍有一系列的缺陷,如导管器材昂贵,病人在整个过程中需要麻醉,具有一定风险,对医生的操作经验要求较高,整个设备对于医生也有一定的辐射风险。因此急需开发基于肿瘤特异的栓塞策略用于治疗。若能使用纳米尺度的生物纳米材料,通过静脉注射靶向递送栓塞剂,引起肿瘤局部血管栓塞,通过调控血液的凝聚态对肿瘤进行治疗,将具有很好的应用优势。
目前研究中,肿瘤栓塞治疗策略的一大障碍是无法将血管栓塞剂特异性的运输至肿瘤血管。而全身性的注射促凝剂会极大限制肿瘤治疗效果,具有严重的不良反应。在过去二十年中,活体内靶向的栓塞治疗方法主要集中在利用抗体或者多肽将组织因子的细胞外结构域(Truncated tissue factor,tTF)选择性的递送到肿瘤。游离状态的tTF可溶且无促凝血活性,当其锚定在磷脂膜附近时,其促凝活性得以恢复。前期已有一些结果显示,tTF的递送策略能够选择性的诱导肿瘤血管栓塞及肿瘤坏死;但由于非特异性运输和快速被网状内皮系统清除,该策略尚未进入临床实践中。pHLIP(pH(low) Membrane insertion peptide)多肽是一种低pH响应型构象变化多肽,在中性溶液中处于自由卷曲状态,在肿瘤组织pH值约6.5的微酸性条件下,可形成α螺旋从而嵌入细胞膜中。这种多肽可以通过静脉给药,利用其酸敏感性而靶向多种实体瘤。基于该多肽良好的肿瘤靶向性,Li等发展了一种肿瘤靶向递送tTF引起凝血的策略[28]。他们构建了一种pHLIP与tTF的融合蛋白,这种融合蛋白与上述策略不同,能够直接通过静脉给药从而靶向运输至肿瘤血管,利用pHLIP的微酸性响应特性而嵌入肿瘤血管内皮细胞膜,恢复促凝功能,在肿瘤区域定点产生凝血。在荷瘤小鼠水平,这种pHLIP与tTF的融合蛋白展示了很好的肿瘤血管特异性凝血的功能,有效减小了肿瘤负荷并避免了其他副作用。
凝血酶是机体凝血系统的一种关键酶。作为一种有效的促凝剂,凝血酶在不需要其他众多凝血因子参与的情况下,快速而高效的诱导血栓的形成。如果将凝血酶作为促凝药物靶向递送到肿瘤血管中,将能够有效阻塞肿瘤血管从而达到治疗肿瘤的目的。然而,凝血酶在血液中的半衰期较短,很容易被清除,其自身的高促凝活性并不适合用于静脉注射。如何实现凝血酶的对肿瘤组织的靶向输送是将其应用于肿瘤治疗的关键挑战。
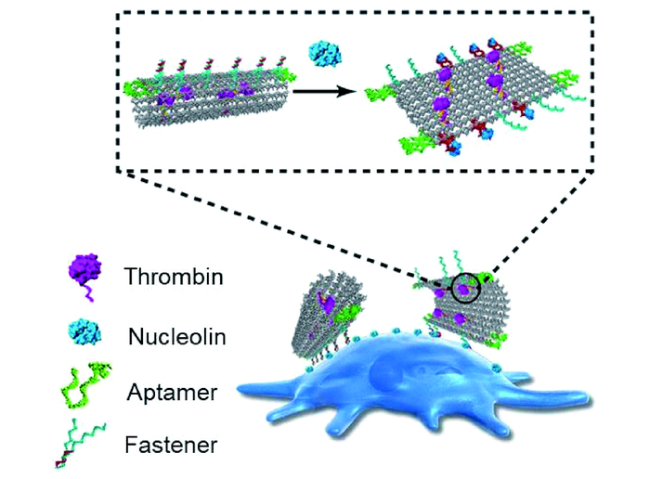
DNA折纸纳米结构具有很好的结构设计特性,可以根据预先需要设计形成确定的纳米结构和形状[16]。Li等利用DNA折纸术设计构建了一个管状DNA分子机器,将凝血酶作为引导血栓形成的药物装载在分子机器内部从而隔绝血液,使其处于非活性状态;两端装载有核酸适配体,利用分子识别提供肿瘤血管靶向识别和定位功能;当DNA分子机器到达肿瘤血管时,其上整合的“分子锁”识别肿瘤血管标志物而发生结构变化,使得“锁”从闭合状态变为开启状态,整个管状结构打开变为平面结构,从而暴露出内部的凝血酶(图3),在肿瘤血管内定点发生凝血产生血栓,通过栓塞肿瘤达到抑制其生长转移的目的[29]。在细胞水平、活体水平,实验结果展示了装载有凝血酶的管状DNA分子机器可以实现活体内的精准运输和定点栓塞,对于包括乳腺癌、黑色素瘤、卵巢癌和原发肺部肿瘤在内的多种肿瘤都有良好的治疗效果。由于DNA分子机器可以实现精确的定位效果,整个体系使用量很低。DNA机器还有很好的识别响应功能,仅在肿瘤血管标志物存在时才开启活化凝血酶,这些性质均保证了装载有凝血酶的DNA分子机器具有极高的特异性,动物实验结果展示了DNA分子机器在小鼠模型和迷你猪模型上都具有良好的安全性。这种智能化的DNA分子机器有望为肿瘤血供阻断治疗策略提供一种高效低毒的药物新剂型。同时,这种DNA分子机器也展示了强大的活体运输与响应识别功能,能够作为智能化的给药平台进行多种药物的高效递送,甚至将促成多种难以成药的物质(如毒素、蛇毒蛋白等)被有效包裹和智能递送,开发出全新的抗肿瘤药物。
3.2 纳米材料对凝血现象的调控——纳米抗凝剂的开发
病理性血栓形成引起的血栓栓塞性疾病,如动脉硬化、深静脉血栓形成、肺栓塞、心肌梗塞和中风,是导致人类死亡的主要原因之一。近年来,由于心脑血管疾病在人群中的普遍高发,抗凝药物已成为现代社会广泛使用的药物之一[30, 31]。例如患有冠状动脉疾病的病人通常表现出急性冠状动脉综合征(Acute coronary syndromes,ACS)症状,需要接受抗凝治疗;如果进行冠状动脉重建,例如搭桥手术(Coronary artery bypass graft,CABG)或介入治疗(Percutaneous coronary interventions,PCI),也需要持续的抗凝治疗[32]。抗凝药物的主要副作用是其引发的出血风险,严重时可导致失能和死亡。应对出血风险及针对性治疗措施是ACS病人治疗中的重大问题。肝素是目前临床使用最为广泛的抗凝药物,对于需要快速起效、全身系统性抗凝的病人可直接用于静脉给药。在临床使用中,肝素仍然面临着较高的出血风险,容易引发肝素抗性、血小板减少(Heparin-induced thrombocytopenia, HIT)等副作用,肝素的解毒剂鱼精蛋白也具有一定的毒性,如过敏反应、肺动脉低血压、心动过缓和消化不良[32]。因此,开发强效、可控、安全的抗凝剂仍然是临床实践的重大需求。
相比于直接清除循环中的抗凝药物,利用解毒剂对于抗凝药物活性进行控制是一种安全的调控方法。核酸适配体是一类经体外筛选得到的短链DNA或RNA片段,可以通过特异性结合靶点蛋白质或酶来调控其功能。一般通过指数富集的配体系统进化技术(Systemic evolution of ligands by exponential enrichment, SELEX),从随机单链核酸序文库中筛选得到与靶点物质具有较高亲和性的核酸适配体序列。目前已有一系列的靶向抑制凝血因子的核酸适配体被筛选出来,用于高效率、解毒剂可控的抗凝剂研究中。Rusconi等[32]筛选得到抗人凝血因子IXa的核酸适配体(aptamer 9.3 t,修饰RNA),体外验证显示其具有很好的抗凝活性。对于该核酸适配体他们首先进行了5’端胆固醇修饰(Ch-9.3 t),以增强核酸适配体在循环系统中的存留时间。他们设计了与该核酸适配体部分互补的序列(5-2C,修饰RNA),通过杂交形成双链,破坏适配体与凝血因子IXa结合域,实现其解毒的功能。研究结果显示,这种具有抗凝作用的核酸适配体能够在模型猪体内实现系统性的抗凝作用,能够在小鼠模型中抑制血栓形成。在活体水平,抗凝剂对应的解毒剂序列能够迅速有效的逆转其抗凝作用,显著降低模型动物的出血现象。结果展示了基于合理设计的核酸适配体在抗凝剂-解毒剂的研发及临床应用中的可能。
虽然核酸适配体已经在现有药物的开发中展示了很好的前景,但仍面临许多问题,如易受到核酸酶攻击而降解,在活体内快速的肾清除等,这些缺点不利于其作为静脉注射药物的应用。核酸序列在纳米材料表面的三维排布能够有效增加其稳定性,增强其对于核酸酶的抗性,将其纳米化可有效延长核酸适配体在血液循环中的存留时间。利用纳米颗粒通过表面修饰具有抗凝作用的核酸适配体,构建纳米抗凝剂,进行凝血酶活性调控及新型抗凝剂开发研究的工作目前已有报道。Wang等[34]利用金纳米棒(AuNR)和磁珠(MB)为核心,共组装一种“闭环”核酸适配体结构,在近红外光作用下,调控凝血酶活性(图4A)。他们采用识别凝血酶两种DNA适配体(TBA15及TBA29,分别结合凝血酶的exosite I及exosite II)形成一个“闭环”结构增加适配体的局部浓度,从而协同增强其与靶点的结合能力。金纳米棒首先被固定于磁珠上(MB/AuNRs),TBA15经延伸和巯基化修饰后通过金-硫键修饰于金纳米棒表面(MB/AuNRs-TBA15),TBA29则延伸出与TBA15延伸序列互补的片段,通过杂交的方式形成Y型的“闭环”结构(MB/AuNRs-Closed-loop)。这种结构将两种凝血酶识别的适配体片段结合后,能够有效增加对试管内凝血酶的捕获结合,从而有效抑制凝血酶的催化功能,抑制可溶性纤维蛋白原向不可溶的纤维蛋白转化。这种蛋白凝聚态变化的动态过程可以直接在激光共聚焦荧光显微镜下观察。金纳米棒由于其特有LSPR性质而具有良好的光热转换效果。利用近红外激光(808 nm)照射时,金纳米棒会将光子转化产生大量的热,从而破坏“闭环”的适配体杂交结构,释放出TBA29。因此,被捕获的凝血酶得到释放,其促凝活性也得到恢复。余下的纳米颗粒结构(MB/AuNRs-TBA15)可以利用磁珠部分纯化并再次利用进行下一轮酶活力抑制试验。该研究工作为研究蛋白质活性调控、分子间识别及其对生物功能的影响提供了一种崭新的策略。
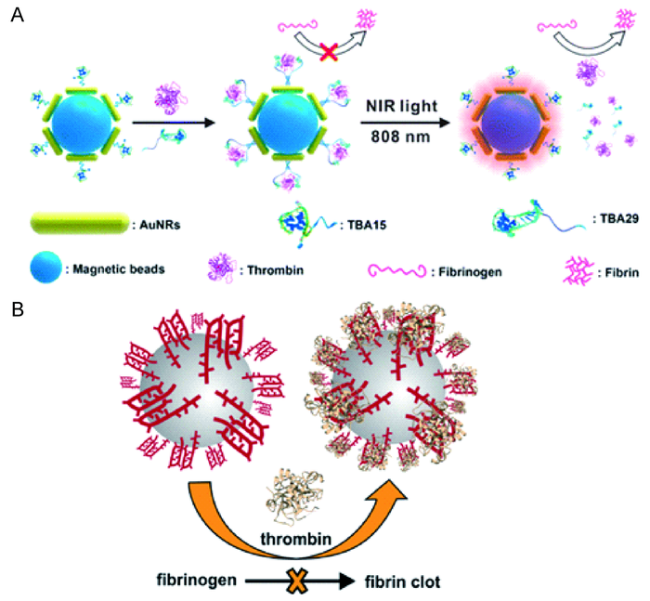
图4 纳米抗凝剂。(A)金纳米棒和磁珠为核心共组装的“闭环”核酸适配体结构[34]及(B)DNA适配体-高分子胶束[37]用于抗凝 Fig.4 Nano-anticoagulants. (A) Thrombin Inhibition by the MB/AuNRs-Closed-Loop Structure [34]. Copyright 2015, American Chemical Society.(B) Self-assembled DAPA-NPs and the thrombin inhibition[37]. Copyright 2017, American Chemical Society |
Huang等[35]利用13 nm的金纳米颗粒作为核心,负载了两种与凝血酶特异识别结合的DNA核酸适配体,开发了一种光响应的纳米抗凝剂。他们首先对金纳米颗粒所负载的两种DNA序列进行设计:末端的聚腺嘌呤(polyA)吸附于金纳米颗粒表面,使得核酸序列能够与金纳米颗粒稳定结合;中间段的核酸用于将两条DNA序列杂交成双链;另一末端则是用于与凝血酶识别结合的片段(TBA15及TBA29)。这种基于纳米颗粒的设计能够使得两种核酸适配体序列具有很好的密度、相对距离与空间取向,增强了纳米抗凝剂与凝血酶之间的相互作用,有效提升了其抗凝作用的效果,抑制了纤维蛋白原向纤维蛋白的转化。对比商品化的抗凝剂(肝素、阿加曲班、水蛭素或华法林),这种基于金颗粒的纳米抗凝剂有效延长了试管内凝血酶引导的凝聚时间。在鼠尾出血实验中,这种纳米抗凝剂的效果也优于肝素。这种基于金颗粒的纳米抗凝剂在人血浆中具有很好的稳定性(半衰期>14 d),具有很好的生物相容性,没有明显的细胞毒性,也不会造成溶血。由于金纳米颗粒的光热转换效应,在该体系受到绿色激光(532 nm,1 W/cm2,3 min)照射时,其热效应将会使得约45%核酸适配体从金纳米颗粒表面脱落,破坏适配体与凝血酶的结合,恢复凝血酶活性,对该体系的抗凝作用产生抑制作用。若采用巯基化修饰DNA适配体直接通过金-硫键偶联至金纳米颗粒表面,则仅有约1%的适配体从金纳米颗粒表面脱落。这种外界光控的方法为控制纳米抗凝剂活性、进一步用于实际的手术伤口愈合提供了一种新颖和安全的抗凝方式。
除了上述基于核酸适配体和纳米颗粒组装的纳米抗凝剂构建方法,Krissanaprasit等[36]利用核酸自组装技术构建了一个具有抗凝功能的单链RNA折纸纳米结构。前述方法中虽然通过闭环杂交的模式将结合凝血酶的两种核酸适配体连接起来,但是这种连接仍然具有很强的柔性,空间控制相对较弱。DNA和RNA自组装结构能够在纳米尺度下精确的引导功能基团进行定位组装,对于控制核酸适配体的空间位置、相对距离具有很强的优势。他们首先利用了计算机设计了RNA折纸结构的形状,并利用体外转录技术获得了带有修饰碱基的单链RNA模板(核心序列210 nt),自组装形成2-螺旋的RNA折纸纳米结构,提供四个位点可供放置RNA适配体。在该单链RNA中包含了两个凝血酶结合的RNA适配体序列(RNAR9D-14T和Toggle-25 t,分别结合凝血酶的exosite I及exosite II),自组装形成RNA折纸结构后,这两个RNA适配体可根据预先设计分别在该结构的四个位点伸出。根据两个RNA适配体排布位置不同,获得了几种RNA折纸结构,包括两种RNA适配体在2-螺旋的RNA折纸靠近的同侧位置,或相对远离的同侧或对侧位置。对于几种不同排布模式的RNA折纸结构进行抗凝活性的测试,结果显示当两种RNA适配体处于距离靠近的同侧位置时,RNA折纸结构具有较好的抗凝活性,血浆活化部分凝血活酶时间(Activated partial thromboplastin time,APTT)较游离RNA适配体有明显的延长。这种RNA折纸引导的纳米抗凝剂也可利用互补DNA序列对其进行解毒,迅速逆转其引发的抗凝活性。由于RNA单链体外转录时使用了2’氟代修饰碱基,由此构成的RNA折纸纳米结构具有较好的血清稳定性,带有RNA适配体的折纸结构在4 ℃放置可超过90 d仍具有较好的抗凝活性。该工作为基于核酸自组装纳米结构的抗凝剂开发提供了新思路。
Roloff等[37]利用凝血酶结合的DNA适配体与高分子片段进行共价偶联,合成得到两亲性分子,并利用其自组装性能开发了一种基于胶束、尺寸可控的纳米抗凝剂(图4B)。这种DNA适配体两亲性高分子(DNA-aptamer polymer amphiphiles,DAPA)可以自发组装成尺寸约10 nm的均一的纳米胶束颗粒(DAPA-NPs),使得每个DNA适配体占据8 nm2的表面积,相比于金属纳米颗粒体系提升了核酸适配体在纳米颗粒表面的分布密度。他们对不同样品进行了血清孵育,证实了这种胶束控制的三维分布的核酸适配体能够有效提升其对于核酸酶的抗性。圆二色性光谱展示胶束上三维分布的核酸适配体保持了具有生物活性的二级结构,在血浆实验中也有效抑制了凝血酶的功能,延长了血浆凝聚的时间。在人血浆活化部分凝血活酶时间(Activated partial thromboplastin time,APTT)及凝血酶原时间(Prothrombin time,PT)检测中,基于胶束的核酸适配体纳米颗粒较游离核酸适配体均有更大幅度的时间延长,展示了更好的凝血抑制功能,展示了核酸适配体三维分布和局部浓度富集对于凝血功能调控的重要作用。利用互补序列与三维分布的核酸适配体杂交,破坏DNA适配体的二级结构,也能够很快的解毒,恢复凝血酶的功能。同时,这种纳米尺寸、可解毒的抗凝剂也显示了更好的活体水平长循环效果,较游离适配体序列(注射后30 min约留存6%)更优,注射后30 min仍具有53%颗粒留存于小鼠活体内。该工作为拓展核酸适配体的稳定性和凝血功能调控提供了新策略。
在血液凝聚态调控过程中,血小板是非常重要的成分之一。除了在血液凝固的生理过程中发挥重要的促进作用,血小板在维持肿瘤血管完整性方面也提供了重要的保护作用,对肿瘤的发生发展均有重要影响。肿瘤微环境中,大量中性粒细胞浸润可引起血管的损伤,削弱内皮连接。血小板可以通过分泌5-羟色胺(5-HT)、血小板第四因子(PF-4)、转化生长因子(TGF)β等颗粒内容物或通过直接黏附于血管受损破裂处,维持血管完整性,促使肿瘤快速生长,并阻止纳米药物向实体瘤扩散。系统性的清除血小板能够显著提升小分子药物的递送,增加化疗疗效,但可能会造成很高的出血风险。在肿瘤区域定点敲除肿瘤相关血小板将是一个打破血管屏障、增加纳米颗粒从血管外渗透的良好策略。Li等[38]构建了一个基于高分子-脂质-多肽的药物递送系统[Polymer-lipid-peptide(PLP)-based drug delivery system]用于定点清除肿瘤相关血小板,实现了高效的肿瘤治疗。这种具有核壳结构的PLP纳米颗粒由两部分组装而成:高分子内核纳米颗粒由生物相容性良好且可降解的嵌段共聚物PEI-(PLGA)2自组装而成,包载小分子化疗药物阿霉素及吸附血小板抗体R300;壳层包含基质金属蛋白酶Ⅱ(Matrix metalloproteinase 2,MMP2)响应多肽序列、卵磷脂和PEG修饰磷脂。基质金属蛋白酶Ⅱ特异的表达在肿瘤内皮细胞和肿瘤基质细胞的质膜上,是一种很有潜力的肿瘤微环境相关的靶点。这种PLP给药体系的壳层在肿瘤微环境被基质金属蛋白酶Ⅱ切割而脱落,在肿瘤微环境中特异性的释放出血小板抗体R300。血小板抗体与血小板表面受体结合,诱导血小板微聚集体形成并定点清除血小板。在肿瘤区域特异性的清除血小板后,肿瘤血管间隙变大,促进了包载药物纳米颗粒高效渗透进入肿瘤内部。这种定点清除血小板的多功能纳米药物体系有效的抑制了乳腺癌和肺癌模型小鼠肿瘤的生长和转移。该工作为利用纳米材料调控肿瘤血管进行治疗提供了实验依据。
3.3 纳米材料对于阿尔茨海默病的诊断与治疗
目前临床上针对于AD的诊断和监测多利用认知行为量表等临床神经心理学方式,较为主观,且着重于疾病的中后期。关于AD的新型检测手段正在开发中,如脑脊液检测和利用正电子断层扫描(Positron emission tomography,PET)。但这些方法仍面临侵入性检测手段(腰椎穿刺)或检测费用高昂的问题。发展基于血液的AD早期检测手段具有很好的临床前景,但仍具有很多挑战。相比于脑脊液,循环系统中病理性的AD分子浓度显著较低,造成检出十分困难。血浆中Aβ水平接近传统ELISA检测的下限,同时血浆中Aβ水平分析与脑内斑块沉积的相关性有限。PET影像技术通常用于确定脑内的Aβ斑块负荷,有限测量不溶性的纤维状沉积物,而基于血液的检测方法则有可能分析血浆中可溶性的Aβ总量。然而,这种检测的差异性提出了一个问题:是否存在能够更好反应脑内病理学特征的循环Aβ亚群。外泌体是一种存在于循环系统中重要的细胞外囊泡,由细胞分泌至循环系统中,尺寸为50~150 nm。外泌体可作为细胞间通讯的信使,可携带多种分子货物跨越生物屏障(如血脑屏障)。有研究显示外泌体与病理性AD蛋白相关,外泌体标志物在脑内淀粉样蛋白斑块中富集,显示外泌体在AD病理学发生和进展方面可能起到了重要作用。捕获这些外泌体相关信息可能对于AD的血液检测以及分子水平确认有较大的意义[39]。
Lim等[39]发展了一种直接从血液中检测循环Aβ蛋白分群的方法。他们使用AD病人的血液样品,提取获得外泌体。他们搭建了一种具有纳米孔阵列结构的等离子体纳米传感器作为基底,捕获外泌体;当外泌体上存在疾病相关分子靶标时,原位酶扩增技术使得外泌体周围迅速产生不可溶的光学沉积物。这种沉积物可用于分子共定位分析,并且折射率改变增强了表面等离子共振(Surface plasmon resonance,SPR)信号。利用这种检测方法,他们发现前纤维Aβ聚集体优先与外泌体结合,因此定义为外泌体结合的Aβ亚群。针对AD病人和对照组人群血液中,外泌体结合的Aβ亚群(Aβ42+CD63+)丰度进行检测,发现与未结合或循环中总Aβ相比,外泌体结合Aβ亚群能够更好的反应脑内淀粉样蛋白斑块沉积,与PET影像学结果有较好的相关性。
自β-淀粉样蛋白级联假说提出至今,已有近三十年历史,虽然该假说在AD药物研发领域仍具争议,但仍然是获得最多科学证据支持的AD致病机理。一系列针对Aβ的治疗方式已被广泛研究,靶向Aβ形成、聚集、清除各个环节的药物研发逐渐成为研究热点,已用于出现临床症状或具有较高风险的阿尔茨海默病人的评价。如一系列抗Aβ的单克隆抗体类药物已进入临床试验阶段,如靶向Aβ寡聚集体及斑块的Bapineuzumab,靶向单体状态的Aβ的Solanezumab,靶向Aβ单体、寡聚体、纤维的Crenezumab等,目前尚未完成评价或已失败[9]。2016年Sevigny等[40]报道的单克隆抗体Aducanumab则可选择性结合Aβ寡聚集体和纤维,在转基因AD小鼠模型结果显示,Aducanumab可进入小鼠大脑中,结合Aβ,剂量依赖的减少可溶性和不可溶的Aβ,清除Aβ斑块。针对轻度AD患者,每月静脉注射抗体持续一年,PET成像结果显示能够有效降低脑内Aβ斑块,结果具有时间和剂量依赖性。经过临床评估认知能力的评分系统,患者认知能力的下降也得到减缓。其临床效果需要更大规模的临床试验确证,目前针对该抗体的临床试验正在进行中,也引发了许多的讨论。
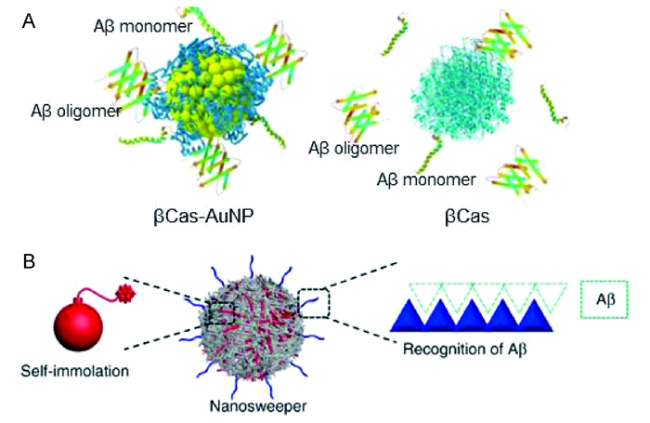
除了常见的单克隆抗体类药物,以纳米颗粒作为Aβ抑制剂的开发也展示出许多优势,如较小的毒性,能够实现长循环,能够跨越血脑屏障,针对毒性的Aβ聚集也具有较好的靶向性,各类工作近年来也有许多报道。β酪蛋白(βCas)是一种乳清蛋白,具有高达18%的脯氨酸残基,同时整个蛋白不含有二硫键,便于蛋白形成开放柔性的构象。单体的酪蛋白多呈无序状态,可在疏水相互作用和静电力驱动下形成胶束。βCas的结构特性使其具有类似伴侣蛋白的活性,如缺乏三级结构和溶剂暴露的疏水性,以异构的低聚物形式存在,存在动态可延展的蛋白质区域,能够与许多部分折叠的蛋白质结合从而防止其聚集等。酪蛋白这种类似伴侣蛋白的行为可以保护淀粉样蛋白生成区域,在天然状态下防止乳腺或乳汁中的αs2及κ-酪蛋白变性,有报道称可以体外抑制胰岛素和Aβ40的淀粉样变性。Javed等[41]利用了一种酪蛋白包被的金纳米颗粒(Casein coated-gold nanoparticles,βCas AuNPs)在模式动物斑马鱼中进行试验抑制Aβ聚集及其产生的毒性。他们利用硼氢化钠还原法合成约5 nm的球形金纳米颗粒,修饰βCas(图5A)。较小的尺寸有利于高效的跨血脑屏障运输,同时保持βCas对于伴侣蛋白活性重要的无规卷曲构象。首先在体外评价了βCas包被的金颗粒在体外对于Aβ42的抑制效果,结果展示其对于Aβ寡聚集体具有很好的亲和力。他们利用心内给药的方式对斑马鱼AD模型进行系统性给药,显示抑制斑马鱼幼鱼和成体脑内的Aβ42聚集引发的毒性,证明了βCas包被的金颗粒对于恢复成体斑马鱼运动能力和认知能力的有益作用。通过非特异性、类似伴侣蛋白的结合作用,βCas包被的金颗粒实现了对Aβ42毒性的抑制。单独酪蛋白的对照组未见明显的毒性抑制作用,可见金纳米颗粒在该体系中的重要作用。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
图5 用于Aβ调控与清除的纳米材料。(A)酪蛋白包被的金纳米颗粒用于增强Aβ亲和作用[41]。(B)基于多肽的自组装纳米颗粒用于捕获和清除Aβ聚集[42] Fig.5 Nanomaterials for Aβ regulation and clearance. (A) Enhanced binding of Aβ with βCas promoted by the AuNP substrate[41]. Copyright 2019, Nature Publishing Group.(B) Peptide nanoparticle captures Aβ by hydrogen-bonded co-assembly and internalizes a substantial amount into cells carrying Aβ[42]. Copyright 2018, Nature Publishing Group |
阿尔茨海默病重要的病理学特征之一即Aβ自组装纤维的细胞外沉积。设计AD疗法时,Aβ的清除是维持体内Aβ平衡的关键。自噬(Autophage)是细胞降解自身代谢物的方式,已有报道显示自噬-溶酶体系统功能障碍会导致Aβ聚集,适当提高自噬对于Aβ的清除有益。然而,Aβ由内含体中淀粉样前体蛋白APP产生,被重循环至细胞表面,通常沉积于细胞外。而自噬作用对物质的降解发生于细胞内,若要尝试利用自噬的方式增加Aβ的清除,首先需要解决的是Aβ的捕获和细胞内化。Luo等[42]设计了一种基于多肽的自组装纳米颗粒,用于捕获和清除Aβ聚集。这种自组装纳米颗粒的核心是一个阳离子壳聚糖核,表面修饰两种多肽(PEGylated-GKLVFF和Beclin-1,TGFQGSHWIHFTANFVNT)。其中,KLVFF能够识别Aβ并通过氢键相互作用与之结合自组装。Beclin-1则可以诱导自噬,引发Aβ降解。聚乙二醇修饰增加了整个纳米颗粒在水溶液里的分散性和生物相容性。该纳米颗粒能够特异性的结合捕获细胞外的Aβ,与之共组装,抑制毒性的Aβ聚集产生。结合后的纳米颗粒可以携带Aβ进入细胞,激活自噬,降解Aβ,最终导致Aβ的清除。细胞和活体水平实验均显示,这种基于多肽自组装的纳米颗粒可以引导高效率的Aβ清除。在细胞水平,可以将细胞活力从60%提升至93%。在活体水平,可将转基因AD小鼠脑内的不可溶Aβ从1539 ng/mg降至914 ng/mg,将可溶性Aβ从585 ng/mg降至190 ng/mg,引起小鼠记忆力的修复(图5B)。上述这些基于纳米颗粒的AD治疗方式利用了其对于Aβ自组装的调控作用,展示了很好的应用前景。
4 结论与展望
本文探讨了一系列生物大分子的凝聚态及其动态变化现象,如凝血现象与阿尔茨海默病,可见生物大分子的凝聚态变化涉及许多重要的生理或病理过程。纳米技术基于原子或分子组装可构建出新的纳米功能器件或具有新生物效应的纳米材料,为研究生物凝聚态、调控凝聚态变化、发展针对疾病的诊断治疗新方法,提供了强有力的研究手段。
通过总结上述研究工作,可见功能化的纳米材料对于生物凝聚态现象的一系列调控作用:通过构建多种功能化纳米颗粒,在肿瘤血管定点激活凝血因子,引发凝血,导致血液凝聚态变化产生血栓,通过栓塞肿瘤达到抑制其生长转移的目的。通过抑制凝血因子活性,调控血液凝聚态变化,在全身或肿瘤区域抗凝,实现安全可控的纳米抗凝剂开发或高效的抗肿瘤药物运输。通过构建纳米传感器或功能化颗粒,实现淀粉样蛋白的捕获和检测或对其聚集状态、清除方式进行调控,提供了针对阿尔茨海默病新颖的诊断和治疗策略。
虽然上述研究工作已有很好的结果,但在针对生物凝聚态调控,开发新颖的肿瘤、血栓类疾病、阿尔茨海默病的诊疗策略方面,仍面临着许多挑战。如何进一步提升促凝纳米材料肿瘤靶向运输效率,提升纳米材料的环境响应性用于快速恢复促凝活性,研究所产生血栓在活体内清除代谢过程,是后续肿瘤栓塞纳米制剂开发中需要注意的问题。针对纳米抗凝剂的研发,如何通过改进序列设计和纳米材料设计,进一步提升抗凝效果并保证安全性,如何发展合理的抗凝剂应用方式将是后续研究中需要考虑的问题。针对阿尔茨海默病及淀粉样蛋白聚集,研究聚集机理,发展针对Aβ形成、聚集、清除不同阶段的功能化纳米颗粒进行疾病的诊断和治疗至关重要。对于生物凝聚态现象及相应的功能化纳米材料的研究有可能推动含有纳米材料(或纳米成分)的药物制剂和载体系统进一步发展,进入临床试验阶段。对于生物凝聚态及其调控进行详细的研究,有望为开发新一代纳米药物提供新思路与新途径。