



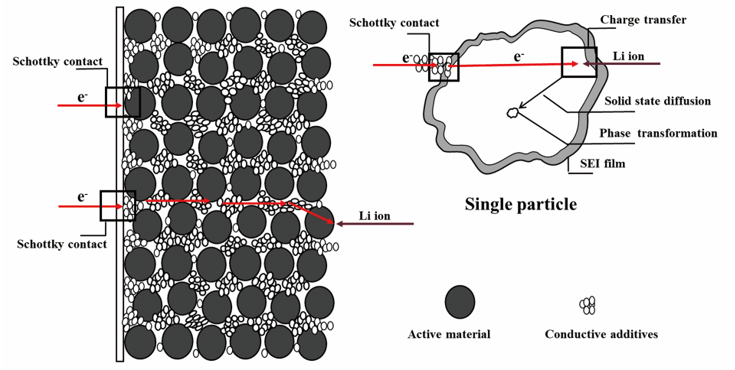
1 引言
自1992年实现商品化应用以来,锂离子电池已在小型移动电子设备如移动电话、笔记本电脑等上得到了广泛的应用,是绝大多数便携式电子产品的电源。近年来,锂离子电池的应用范围已经逐渐扩大到电动汽车和储能系统方面,并有望在军事、航天等领域获得进一步的应用。最近,瑞典皇家科学院将2019年诺贝尔化学奖授予约翰·古迪纳夫、斯坦利·惠廷厄姆和吉野彰,以表彰他们在锂离子电池研发领域作出的贡献,显示了人们在锂离子电池领域已取得的巨大成就。然而,锂离子电池要大规模应用于这些新兴领域仍面临着许多障碍,如何进一步提高其能量密度、循环稳定性和安全性及进一步降低成本等仍然是锂离子电池领域面临的重大挑战。毫无疑问,上述问题的解决依赖于人们对于锂离子电池电极和电解液材料以及其中发生的基本物理化学过程认识上的提高,特别是电极薄膜的厚度、多孔性及其制造工艺(包括电极活性材料与黏合剂、导电剂的比例,电极浆料的搅拌混合、涂布、烘干以及辊压过程等)等对电池性能的影响。电化学阻抗谱(Electrochemical impedance spectroscopy,EIS),早期也称交流阻抗谱(AC impedance),是以小振幅的正弦波电势(或电流)为扰动电信号,使电极系统产生近似线性关系的响应,测量电极系统在很宽频率范围内的交流电势与电流信号的比值(此比值即为系统的阻抗),以此来研究电极系统的方法。作为一种经典的电化学研究方法,EIS具有以下几个方面的优点:(1)EIS能够根据电化学反应中发生的基本物理化学过程的弛豫时间常数的不同,在较宽的频率范围内对不同的基本物理化学过程实现同时表征;(2)作为一种线性的研究方法,EIS的数据处理比较简单;(3)EIS能够实现对电极反应的原位测试和对电池实现在线测试,测试方法简单易行,易于在工业化生产中获得应用;(4)EIS测试实验中一般不需要独特的实验技能和方法;(5)商品化的电化学工作站或综合测试仪一般都具备阻抗测试功能,仪器、设备廉价,一般不需要辅助部件;(6)EIS测试过程中,小幅度的交变信号不会使被测体系的状态发生改变,能够实现无损检测。因此在过去的20多年里,EIS被广泛应用于锂离子电池研究和生产领域,包括研究嵌锂反应机理和容量衰减机制[1, 2, 3,4],测定相关电极过程动力学参数和电池的健康状态(State of Health, SOH)[5,6]与荷电状态(State of Charge, SOC)[7]以及电池的内阻,探讨影响锂离子电池电极性能的相关因素等[8,9]。
然而,EIS在锂离子电池领域的进一步应用还存在一些比较严重的限制,主要包括:(1)EIS实际应用中面临的一个首要问题是其不确定性,主要表现为:一方面,EIS谱特征通常由两种元素组成,即半圆和斜线,很多不同的物理化学过程或一个复杂过程的不同步骤,在EIS中往往具有相似的谱特征;另一方面,当时间常数相近时,不同的物理化学过程或一个复杂过程的不同步骤的EIS谱特征可能会相互重合,成为一个谱特征,导致对电极反应相关的复合阻抗谱的解释比较困难[10]。因此,锂离子在嵌入化合物(简称“嵌合物”)电极活性材料中嵌入和脱出过程的EIS谱中各时间常数的归属一直存在不少争议。(2)锂离子电池中的电极是一个复杂的多孔结构,EIS测试结果不仅受到活性材料本身性质(结构和颗粒大小等)的影响[11],也受到电极制备工艺[12,13]、实验条件(电解池结构以及对电极和参比电极的种类、位置、几何形状等)等的影响。导致基于不同实验方案的实验结果之间的可比性较差,而又不存在完善的标准体系[14,15]。(3)EIS测试中在高频谱和低频谱测试中依然存在一些硬件方面的技术难题,尤其低频谱测试时间较长,仪器测试精度不高。(4)运用等效电路进行处理EIS谱数据时,需预先假定电化学过程的反应机制,同时为阐明上述各种因素对EIS谱特征的影响,需要建立能够合理准确解释嵌入化合物电极阻抗行为的可靠全面的微观数学模型,因此研究者需要具备一定的数学功底和建模能力。
在前文中[16,17],作者从分析嵌合物电极的EIS谱特征入手,探讨了EIS中各时间常数的归属问题,重点讨论了与锂离子在嵌合物电极中嵌入脱出过程相关的动力学参数,如电荷传递电阻、活性材料的电子电阻、扩散以及锂离子扩散迁移通过固体电解质相界面膜(SEI膜)的电阻等,对电极极化电位和温度的依赖关系。但限于当时作者对EIS在锂离子电池研究、生产领域中应用的认识深度的限制,许多重要的问题如具体到每一种电极活性材料的EIS谱特征、接触阻抗和感抗产生的机制、锂离子电池多孔电极理论及阻抗谱数值模拟的原理与建模方法等,在该文中没有涉及或缺乏深入的探讨。因此在本文中,作者在总结课题组近10年来在这一领域的研究工作以及相关文献的基础上,对上述问题进行了深入探讨。
2 锂离子电池电化学阻抗谱分析的理论基础
锂离子电池的充放电是通过锂离子在正负极间的脱出和嵌入来实现的。充电时,正极中的锂离子从基体中脱出,嵌入负极,同时电子从正极活性材料中脱出进入外电路;放电时,锂离子从负极中脱出,嵌入正极,同时正极活性材料从外电路获得电子,使电荷达到平衡。因而,正常的充放电过程中,需要锂离子和电子的共同参与。但锂离子电池的正极活性材料多为过渡金属氧化物或者过渡金属磷酸盐,它们的导电性都不尽如人意,往往都是半导体甚至是绝缘体,导电性较差,必须加入导电剂来改善其导电性;即使是导电性较好的负极材料如石墨,如果在电极中不添加导电剂,也会严重影响其容量发挥和倍率性能。同时,电池在使用过程中要进行多次充放电,活性颗粒经历周期性的膨胀收缩,颗粒间的接触减少,间隙增大,甚至有些颗粒发生物理脱离,不能再参与电极反应,所以也需要加入导电剂以提高电极结构的稳定性。而黏合剂是粘结电极活性物质、导电剂和集流体的介质,主要功能是将电极活性材料、导电剂和集流体粘结在一起,使电极活性材料之间在保持一定孔隙率的前提下具有整体的连接性,保持活性物质、导电剂和集流体间的接触完整性,避免活性物质颗粒在电池充放电过程中粉化、脱落,使电池始终保持优异的电化学性能[18,19,20]。因此,锂离子电池电极一般是由活性材料、导电剂和黏合剂组成的一个复合电极,导电剂在其中形成的导电网络的性能严重影响着电池的内阻,对电极性能的发挥具有举足轻重的作用[21,22]。
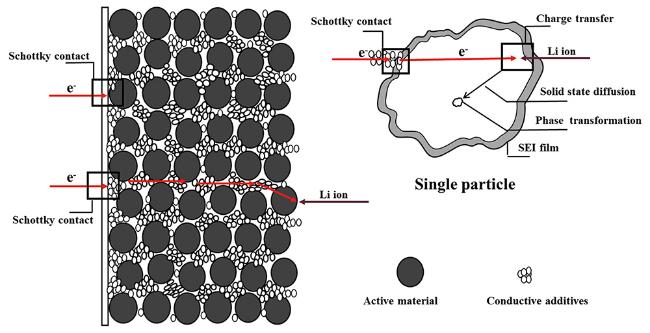
可以看出,电子的输运过程至少包含6个步骤:(1)电子从外电路输运到集流体;(2)电子从集流体传输到导电剂;(3)电子通过导电剂输运到导电剂与活性材料的结合处;(4)电子从导电剂传输到活性材料(即电子从导电剂的导带跃迁到活性材料的价带);(5)电子在活性材料内部的输运过程(通常与活性材料的电导率有关);(6)电子直接从集流体输运到与集流体直接相接触的活性材料的过程。在上述6步电子输运过程中,由于集流体、导电剂和活性材料三者之间的导电性能不同(或者说是功函数不同),因而(2)、(4)和(6)步骤有可能会形成所谓的肖特基接触,也就是金属-半导体接触问题,即在集流体/导电剂、导电剂/活性材料、集流体/活性材料界面处半导体(导电性差的一侧)的能带弯曲,形成肖特基势垒,势垒的存在会导致大的界面电阻。虽然集流体与导电剂一般都具有较好的导电性能,它们之间功函数差距不大,有可能形成欧姆接触,但集流体表面一般都存在金属氧化物保护膜,或由于电极膜与集流体之间的粘结力不够,接触不够紧密,导致二者之间界面形成较大的势垒。在Al或Cu集流体上包覆炭层或去除集流体上的表面氧化层或对集流体进行表面疏水性修饰增进电极膜与集流体之间的结合力,都被证实能够显著降低界面电阻,改善电池的充放电性能[24,25,26]。因此,电子输运过程的(2)、(4)和(6)步骤在EIS中并不只是简单表现为欧姆电阻,而是表现为与电阻和电容并联相关的半圆。
锂离子的输运过程则至少包含以下4个步骤:(1)锂离子在电解液本体中的扩散,通常表现为欧姆电阻,即在EIS中呈现为高频区域与实轴相交的一个点;(2)锂离子的溶剂化/去溶剂化过程;(3)锂离子在多孔电极中电解液的扩散;(4)锂离子通过覆盖在电极表面的SEI膜扩散。此外,如果电解池中存在隔膜则还要包括锂离子在隔膜中电解液的扩散过程。
电化学反应过程,即在电极/电解质界面的电荷转移反应及其后续步骤(质量输运过程,例如浓度梯度驱动的扩散),主要包括4个步骤:(1)电荷传递过程,即完全去溶剂化的吸附锂离子进入活性材料晶格,同时电子进入嵌锂位附近活性材料的价带,使电荷达到平衡;(2)锂离子在电极材料内部的固态扩散,这一过程与电子的扩散是耦合的,也就是说锂离子的固态扩散同时必然伴随电子的扩散,这与单纯的锂离子扩散(如在电解液中的扩散)是完全不同的;(3)锂离子和电子在电极材料颗粒内部的积聚;(4)相变,锂离子和电子在电极材料颗粒内部的积聚导致新相的生成,当新相与电极材料本体物理化学性质差别较大时,则产生相变,因此相变往往伴随活性材料晶体结构的改变、体积的膨胀或收缩以及活性材料颗粒内部应力的产生。
电化学阻抗谱研究的目的就是探明在锂离子嵌入脱出过程(电极极化过程)中何种阻力处于主导地位,即决定电池内阻的关键步骤是什么以及在长期的充放电循环过程中每种阻力增长的趋势,给出影响锂离子电池电化学性能(如,倍率、循环稳定性和容量等)的关键因素,进而提出改进电池电化学性能的方法,同时给出可用于分析SOC、SOH和SOS的电化学参数。
2.1 肖特基接触阻抗问题
其中A为肖特基接触面积,为热电子发射的有效理查逊常数;为势垒高度,k为玻耳兹曼常数,T为绝对温度,E为外加偏置电压(导体端高电势为正),n为理想因子。
根据欧姆电阻的微分形式:可得到如下关系:
当相互接触的导体和绝缘体没有变化时,因外置偏压较小,可认为不变。则式(2)可变为:
为常数。再对式(3)取对数得:
即接触电阻的自然对数与外加偏置电位呈线性关系。
我们实验中获得的NiF2/C复合材料电极在首次充放电过程接触电阻lnR随电极电位变化,与公式(4)指示的结果相一致[31],转化反应发生前后,接触电阻的自然对数与电极极化电位均呈现较好的线性关系。根据放电过程中曲线的斜率,从式(4)反推出n=353.4,远远偏离理想值1。因此,此时接触处的电流输运可能不仅仅是热电子发射。
考虑到导电剂与活性物质晶格失配,材料中存在较多缺陷,综合考虑电场、隧穿效应、中间层以及载流子在空间电荷区的复合等各种因素,肖特基接触的I-E特性可用下式表示[32]:
当时,式(5)可简化为:
则接触电阻可表示为:
其中式(7)两边取对数得到:
该结果显示接触阻抗的自然对数仍然与偏置电压呈线性关系,但斜率与式(4)相比发生了变化。通过对实验数据的线性拟合值反解出此时n=0.998,非常接近1,显然式(8)更适合于描述锂离子电池中的接触阻抗。
2.2 感抗产生的机制
在电路中,当线圈中有电流通过时,就会在线圈中形成感应电磁场,而感应电磁场又会在线圈中产生感应电流来抵制通过线圈中的电流。因此,我们把这种电流与线圈之间的相互作用称为电的感抗,也就是电路中的电感(L),单位是“亨利(H)”,以美国科学家约瑟夫·亨利命名。交流电路的感抗表示电感对正弦电流变化的反抗作用。在纯电感交流电路中,电压有效值与电流有效值的比值称为感抗。用符号XL表示,单位为欧姆,即:
式(9)表明,感抗的大小与交流电的频率f(Hz)及线圈的电感有关。当频率一定时,感抗与电感成正比;当电感一定时,感抗与频率成正比。
在运用EIS研究锂离子电池时,其谱特征中经常会出现与感抗相关的成分,通常表现为Nyquist图中一个开口向上的半圆或一段实轴以下的弧线。感抗按其出现频率范围大致可以分为高频感抗(>104 Hz)、中频感抗(约100 Hz附近,在Nyquist图中一般出现在与锂离子通过电极活性材料表面的SEI膜扩散迁移过程有关的半圆和与电荷传递有关的半圆之间)和低频感抗(<1 Hz,在Nyquist图中一般出现在与电荷传递有关的半圆和反映锂离子固态扩散过程的斜线之间或直接出现在低频区域)。
高频感抗一般认为是由于电极的多孔性、导线电感和电极卷绕电感产生的[33],往往只在阻抗很小的体系能够被明显地观测到,这一电感和电池等效电路的其余部分之间为串联关系。在商品化锂离子电池中,极片被卷成一个层层包裹的卷芯,本身就是一个线圈,高频感抗的出现几乎都是必然的,但一般很难会形成一个完整的开口向上的半圆。在实验室研究中,由于一般都是使用单一极片,高频感抗往往不会出现。此外,所有电路元件既不是纯粹的电阻性元件,也不是纯粹的电抗性元件,而是这些阻抗成分的组合。当寄生电容(Stray capacitances)与电阻相结合时,在阻抗谱中就有可能出现与感抗相关的一个完整半圆,并对测试频率呈现较强的依赖关系[34]。
一般认为,中频和低频感抗与电化学的反应机制有关。在电催化反应中,当连续异相反应中存在吸附中间体时,由于吸附在电极表面的反应中间体的催化效应降低了反应电阻,阻抗谱中就会出现与感抗相关的半圆[12, 35]。然而,感抗是由吸附中间体的催化效应引起的这一观点,很难用于解释锂离子电池中出现的感抗行为,特别是石墨电极EIS中出现的感抗行为。在锂离子电池研究领域,感抗行为产生的原因主要有3种:(1)感抗是由测量过程中活性材料发生相变引起的[36];(2)感抗是由于电极中不同活性材料颗粒之间嵌锂度不同,形成浓差电池引起的[37];(3)感抗是由于电极活性材料表面SEI膜不稳定引起的,即阻抗测量过程中SEI膜的分解、再形成造成的[38]。此外,实验中参比电极摆放的位置不当,例如将参比电极摆放在研究电极或对电极边缘处,会造成EIS中出现与感抗相关的半圆,如果将参比电极摆放在研究电极或对电极的外面,会使与感抗相关的半圆尺寸变大[39],这属于实验技术问题,不在本文讨论范围内。
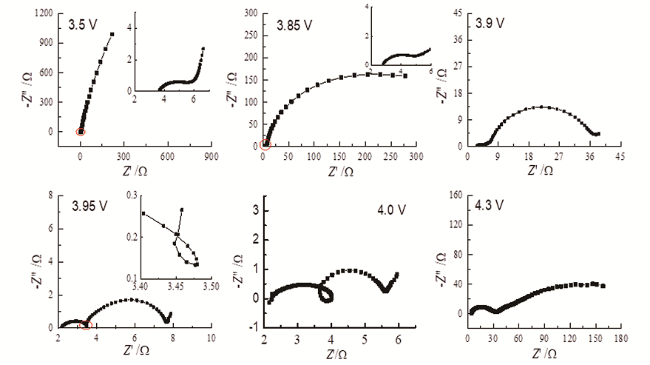
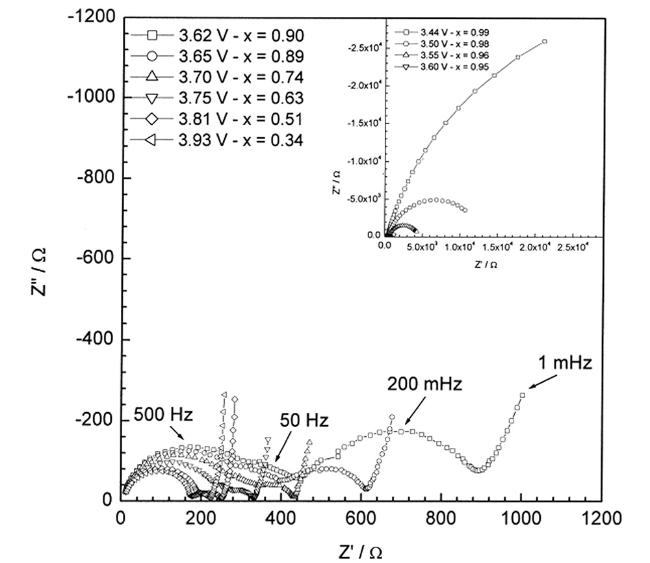
我们研究发现,当电极中导电剂含量较大时,EIS中基本不会出现与感抗相关的半圆,然而当电极中导电剂的含量较少时,则EIS谱特征中容易出现与感抗相关的半圆。图2为我们运用EIS研究的按质量百分比为92%的活性材料、5%的PVDF-HFP黏合剂和3%的导电炭黑组成的LiCoO2电极的首次充电过程[40]。研究发现,与感抗相关的半圆(IL)首先出现在3.95 V(半圆出现在与锂离子通过电极活性材料表面的SEI膜扩散迁移过程相关的半圆和与电荷传递有关的半圆之间,为与中频感抗相关的半圆),即发生部分锂离子脱出的电位区域。当电位高于4.3 V时,即锂离子全部脱出后(对应于LCoO2, x=0.5时),IL消失。因为活性材料在锂离子脱出/嵌入过程中无论是相变的发生还是SEI膜的稳定性一般与导电剂含量没有直接联系,同时对LiCoO2电极而言,4.3 V以下不存在剧烈的相变过程,因此我们的实验结果显然支持感抗是由于电极中不同活性材料颗粒之间嵌锂度不同,形成浓差电池引起的。
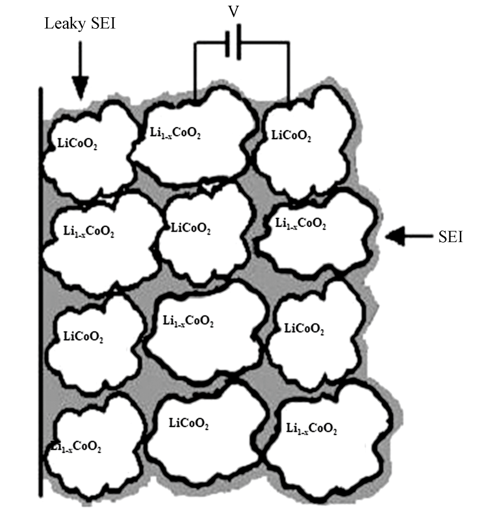
图3中给出了LiCoO2电极中LiCoO2/Li1-xCoO2局域浓差电池模型的示意图。在LiCoO2正极充电过程中,由于锂离子脱出过程中的不均衡性,导致不同的LiCoO2颗粒之间锂离子的脱出量不同,产生被SEI膜分隔开的富锂区域和贫锂区域,即最简单的情形是在被SEI膜分隔开的LiCoO2颗粒和Li1-xCoO2(0<x<0.5)颗粒之间形成局域浓差电池。同时SEI膜并不是完美无缺的,在锂离子脱出过程中,LiCoO2/Li1-xCoO2局域浓差电池中的两电极之间就会有局域电流通过,从而产生电场对抗锂离子脱出产生的电场。伴随锂离子的脱出,LiCoO2/Li1-xCoO2局域浓差电池会不断地渗漏电流,直至锂离子完全脱出(对应于LiCoO2全部转变为Li0.5CoO2),即LiCoO2正极内部不再存在锂离子的浓差极化,此时感抗消失,这一理论预测与我们实验中获得的结果也相一致。
进一步,我们运用EIS研究了实验室合成的尖晶石LiMn2O4电极的首次充放电过程[41],发现3.975 V 时EIS中分别在100 Hz和1 Hz附近各出现一个与感抗相关的半圆(即与中频和低频感抗相关的半圆),这与锂离子在尖晶石LiMn2O4材料中嵌入和脱出过程是分两步进行的相一致,同样表明感抗是由于存在局域浓差电池引起的,这两种局域浓差电池可分别表示为LiMn2O4/Li1-xMn2O4和Li0.5Mn2O4/Li0.5-xMn2O4。当电极极化电位高于4.175 V时,与低频感抗相关的半圆消失。此外,我们还观察到在第二周充放电循环过程中感抗几乎完全消失,进一步表明,感抗是由于存在局域浓差电池引起的[42]。
2.3 锂离子电池多孔电极理论与阻抗谱数值模拟
多孔电极是由高比表面积的粉末状活性物质或与具有导电性的惰性固体微粒混合,通过压制、烧结或粘结等方法构成的电极。多孔电极具有较大的孔隙率,可以大大提高电极的真实表面积,减小工作时的真实电流密度,降低电化学极化。同时,可减小电流流经的距离,减小欧姆电位降,有利于减少电池的能量损失。多孔电极收集到的总电流是三维体系中局部位置上电极反应、扩散和对流等不同程度的叠加结果。因此,发展多孔电极理论并用于电化学能量转换器件,特别是锂离子电池的分析,一直都是个极具挑战性的问题。
在多孔电极中,电极反应是在三维空间结构中进行的,电极结构对电极反应具有重要的影响,直接决定着电池的性能。在电极活性物质一定的情况下,电极结构包括电极组分、组分分散状态等方面以及表征电极结构的有关参数。电极组分的分布状态是影响电极整体性能的重要因素,如果电极组分分布不均匀,电极各点的导电性能将会产生差异,一旦发生极化,电极各处的拓扑电位会由于欧姆压降不同而不同,一部分活性物质得不到利用,一部分活性物质则会过充过放,最终导致其活性衰减,容量降低[43]。
表征多孔电极结构的有关参数主要包括:(1)电极厚度。(2)电极孔隙率。是指电极中孔隙体积与其表观体积的比率。(3)孔的形态。多孔体中的孔分为三种,即通孔、半通孔和闭孔,这三种孔在电池充放电过程中起不同的作用。(4)比表面积。单位表观体积多孔电极涂层所具有的总表面积。(5)孔径及孔径分布。多孔体中孔的大小总不相同,为了全面了解多孔体的结构,还需要了解孔径的分布,即不同直径的孔所占的比例。(6)孔隙曲折系数[44]。
多孔电极工作时,其内表面往往并不能均匀的实现电化学反应。孔隙内液相的传质阻力在多孔电极内部产生浓度极化,导致电极内部各点“电极/电解质”界面上极化不均匀,部分抵消了多孔电极比表面积大的优点[45]。研究多孔电极的主要目的是分析这种电极的基本电化学行为,并找出优化其电极性能的方法。因此,多孔电极理论中的主要问题是确定电极过程在多孔介质中的深度分布,探讨影响多孔电极特征电流的主要因素。
理论上,多孔电极一般采用两相多孔电极来描述。两相多孔电极的理论处理主要有两种途径:(1)宏观均一模型,即假定整个多孔体系是由空间互补的两个均匀的各向同性的连续介质组成的,其中一个是固体介质(即电极的基体),另一个是渗入孔中的溶液。在这个模型中,多孔电极的电导率和扩散系数等性质被视为固体介质和溶液的加权平均值。(2)分散孔模型,即假定多孔电极是由彼此不相交联的许多单孔的组合,因而宏观电流是所有单孔中电流的总和,平板上的动力学规律被直接应用于单孔的情况。历史上,分散孔模型首先为多孔电极行为提供了理论解释,迄今对发展两相多孔电极理论仍具有重要作用。而宏观均一模型更便于电化学工程应用,被广泛应用于化学电源的模拟。
电极是电池的核心,是电池内部进行电化学反应的区域,电极对电池性能具有重要影响。锂离子电池电极是由活性材料、导电剂、黏合剂组成的典型两相粉末多孔电极,为了对锂离子电池内部多孔电极性质这样一个复杂的问题实现理论上的分析,建立基于多孔电极理论的锂离子电池电化学模型是解释锂离子电池电化学行为的重要手段,特别是那些难以通过实验研究的影响锂离子电池电化学行为包括阻抗行为的因素。
两相多孔电极的宏观均一模型是由Newman等首先提出的,该模型不考虑孔的真实结构,而把多孔电极视为固体基质和电解液两种连续介质的叠加,即假定,电极区域内任意一点都包含两相,而所有函数都是时间和空间上的连续函数、所有函数都是两相中两个连续函数集的叠加。在给定的时间内,孔内的电极反应速率可能随不同空间位置而变化,反应速率的分布依赖于固体基质的物理构造和电导率、电解液的电导率以及表征电极过程的各个参数。为了对这个复杂的问题进行理论分析,必须建立一个能够解释实际电极的主要特征但又不必精确几何细节的模型,而且这个模型应能用简单测定得到的合适物理参数进行描述。为此,必须对所需的各种平均物理量进行定义,这些平均物理量应是在比整个电极尺度小但比孔结构尺度大的范围内的平均值。例如,具有随机结构的孔材料可用孔隙率(ε)、单位体积的平均表面积(a)和体积平均的电阻率等表征;类似地,也可用体积平均的电导率表征孔隙中电解液相的性质。基于多孔电极理论均量假设并结合锂离子电池电化学机理模型[48],分别给出描述法拉第过程(电荷传递过程)和双电层电容充放电过程、多孔电极内部的传荷与传质过程的方程[49,50],即采用欧姆定律描述固体电势分布,Butler-Volmer动力学方程来描述正负电极与电解液界面的电化学反应过程,采用Fick定律描述电极内部锂离子扩散过程,即可实现对锂离子电池进行数值模拟。多孔电极理论由于忽略了电极内部的真实几何性质,大大降低了数学建模的复杂度,因而在锂离子电池以及燃料电池模型中得到了广泛应用。
目前由Doyle等[51]开发的锂离子电池多孔电极准二维模型(P2D)被广泛用于描述电池内部的电化学反应过程[52,53],并应用于电池仿真和电池设计等领域,是其他更复杂的电池机理模型的基础。然而,锂离子电池P2D模型是由一组复杂的耦合非线性偏微分方程组成的,所以,求解电化学模型需要大量的计算时间和较大的存储内存。当进一步考虑与SEI膜相关副反应时,副反应的偏微分方程与主反应的偏微分方程相互耦合,求解电池内部各处的副反应速率需要求解更加复杂的偏微分方程组,因此一般在建模过程中会忽略电极表面反应过程(即SEI膜的存在)。此外,一方面P2D模型中存在大量的可调参数,另一方面P2D模型中忽略了活性材料颗粒粒度分布、多孔电极的几何结构等因素。因此,求解获得的阻抗谱的各时间常数往往物理意义不明确,与实验中获得的结果不一致。
(1)单个嵌入化合物颗粒阻抗模型的建立
假设每一个嵌入化合物颗粒表面都被一层组分相同、厚度均匀的SEI膜所覆盖,嵌入化合物颗粒与SEI膜之间的界面、SEI膜与电解液之间的界面均形成双电层的结构,如图4所示。因此建立单个嵌入颗粒模型过程中,必须要考虑电荷传递过程和双电层充电过程要通过上述两个界面,同时还要考虑到SEI膜对电子绝缘的特性。
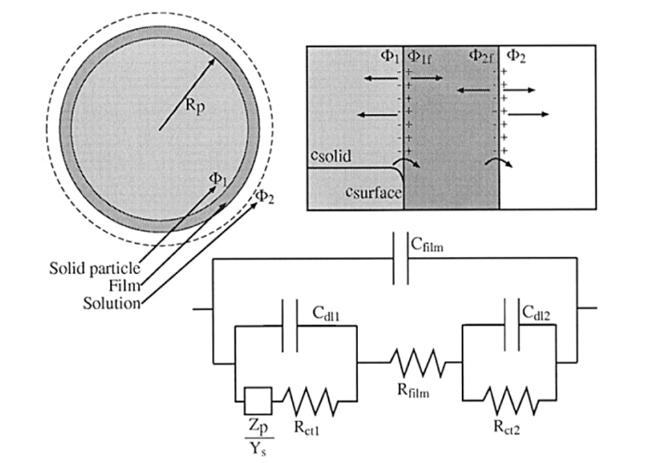
假定在任一界面法拉第电流密度与通过界面的电压降之间的关系符合Butler-Volmer方程:
式中分别为靠近颗粒表面的固相电势和靠近SEI膜表面的液相电势;i0为交换电流密度;F和R为法拉第常数和气体常数;U为电荷传递反应的开路电压;T为热力学温度;αa和αc分别为电化学反应对称因子。为表面过电位,指示了界面偏离平衡的程度。
双电层电容充放电电流密度可表示为:
假定法拉第电流密度和双电层充放电电流密度之间是独立的[59],彼此不产生互相影响,则通过界面的总电流密度可表示为:
电流流经SEI膜的电压降可表示为:
SEI膜电容充放电电流密度可表示为:
SEI膜电容可能非常小,但如果它是电介质,电流就有可能通过两面的分离电荷通过它。因此,只有在很高频率下,才会形成SEI膜电容明显的充放电电流,导致SEI膜短路。
总向外法向电流密度可表示为:
对于传统电极例如金属电极而言,应用式(10)~(15)就可直接求解其阻抗的数学表达式(对嵌入化合物颗粒与SEI膜之间的界面、SEI膜与电解液之间的界面都应用式(10)和(11))。然而,嵌入化合物颗粒的开路电位会随其中锂离子的浓度(嵌锂度)而改变。因此,我们必须通过物料平衡去求解开路电位随电流通过的变化。
如果假定电子迁移率远大于锂离子的迁移率,锂离子在嵌入化合物颗粒中的扩散过程符合Fick定律,那么:
Nintercalant是锂离子的扩散通量,Ds是锂离子在嵌入化合物颗粒中的固态扩散系数,cs是锂离子在嵌入化合物颗粒中的浓度。
在球坐标体系下,由扩散通量引起的物料平衡可表示为:
其中r为径向坐标位置。
嵌入化合物颗粒中表面浓度梯度与界面法拉第电流密度之间的关系可表示为:
考虑到,嵌入化合物颗粒中锂离子浓度在小振幅的正弦波电势(或电流)扰动电信号作用下,随扰动信号正弦波电势(振幅和频率)的变化,式(17)可改写为:
求解式(19)可得:
其中
嵌入反应的开路电位U只与嵌入化合物颗粒表面锂离子浓度有关,为了表达方便,定义嵌入化合物颗粒表面浓度cs为
从而嵌入化合物颗粒表面锂离子浓度与界面法拉第电流密度之间的关系可表示为
定义无量纲传递函数Ys为
将式(22)和(23)代入描述嵌入化合物颗粒与SEI膜之间界面的Butler-Volmer方程,同时对Butler-Volmer进行线性化处理:
定义以下参数:
则法拉第电流密度和通过界面的电压降之间的关系可表示为:
从而法拉第反应的阻抗可表示为:
对于SEI膜与电解液之间的界面而言,由于SEI膜的开路电位不会随其中锂离子浓度的改变而改变(一般情况下,SEI膜中锂离子浓度不会改变),法拉第电流密度和通过界面的电压降之间的关系可表示为:
联立式(13)、(14)、(27)和(29)得,单位界面的总电流与固液相之间的电压降(或电位差)可表示为:
单个嵌入化合物颗粒的阻抗即可由求得。
(2)多孔电极阻抗模型的建立
在获得单个嵌入化合物颗粒的阻抗表达式后,就可以利用它作为传递函数来描述多孔电极,从而液相电流可表示为:
其中为每单位体积电极中半径为的嵌入化合物颗粒数,是每个颗粒的嵌入化合物表面积,是某一粒径嵌入化合物颗粒的每单位表面积的法向电流。如式(30)所示,是固液相之间电压降的函数,定义为嵌入化合物颗粒每单位表面积的外向法向电流密度与固液相之间电压降的传递函数,可以得到:
$\alpha=4 \pi \int_{0}^{\infty}N(r)r^{2}dr$
$\varepsilon_r =\frac{4}{3} \pi \int_{0}^{\infty}N(r)r^{3}dr$
其中a为单位体积电极内包含的颗粒的界面表面积, $\varepsilon_r$为复合多孔电极中固相的体积分数,则
其中为复合电极中所有颗粒的平均导纳。
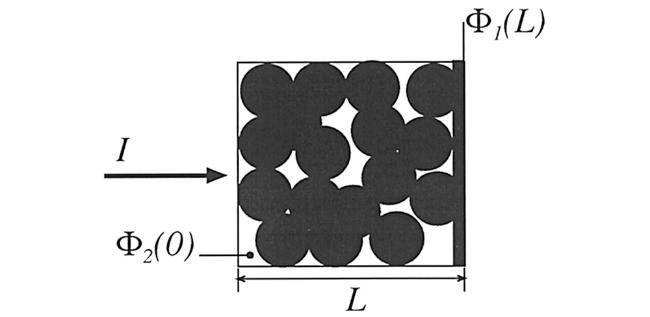
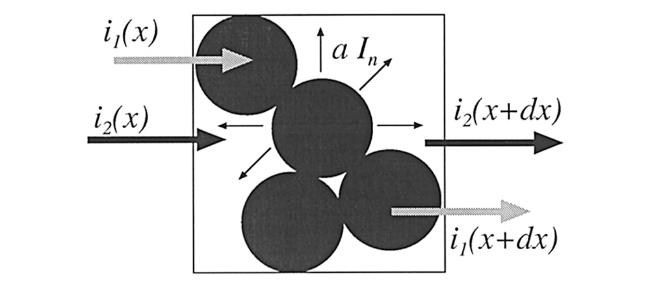
假定在多孔电极中,液相电流、固相电流与电压降的关系均符合欧姆定律,且总电流I等于液相电流与固相电流之和,因而可得到:
将式(35)代入式(36)~(38),同时使用电极的厚度L对坐标x进行无量纲化处理,可得:
显然,上式符合以下边界条件 :
即液相电流不能通过集流体(在集流体/多孔电极界面处的固相电流等于总电流)以及在隔膜/多孔电极界面处的液相电流等于总电流。求解式(39)可得:
其中:
从而获得了多孔电极阻抗的数学表达式。在一些文献中[60],直接用替代用于模拟多孔电极阻抗。
(3)混合嵌入化合物颗粒的总阻抗
i代表某一粒径的嵌入化合物颗粒。商品化锂离子电池一般由典型的两种粒径不同大小的晶粒组成(两个粒径范围),作近似处理,假设多孔电极中只存在两种不同粒径的颗粒时,混合颗粒的总阻抗可表示为:
θ1为颗粒1的阻抗值占混合颗粒总阻抗值的百分比。则进一步可得到:
其中a为所有颗粒的表面积与体积的比值。
从式(41)可以看出,极片厚度是表征多孔电极最重要的参数,对电极的电化学性能有着举足轻重的作用。然而,在实验室中电极片的制备一般都是采用人工涂布的,可能会经常出现的一个现象是所制备电极片的厚度不均匀,即使是工业生产中采用机械涂布时,为提高电池的比能量,需要制备的电极片厚度较厚时,要实现所制备电极片在大尺度范围内厚度均匀,亦是具有一定挑战性的难题。主要因为电极制备过程中,均匀的湿浆料涂敷在金属集流体上,然后通过干燥去除湿涂层中的溶剂。尽管湿浆料中的固含量一般大于30%,但是干燥过程中,溶剂蒸发时,涂层总会经历一定的收缩,才能形成多孔的干燥电极结构。同时,干燥过程中涂层有流平过程,影响涂层的均匀性。
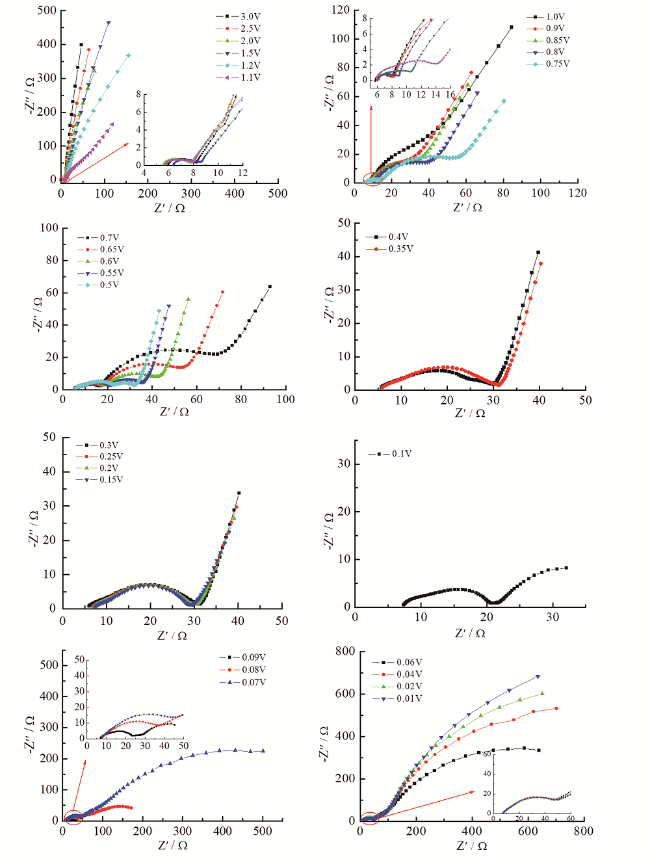
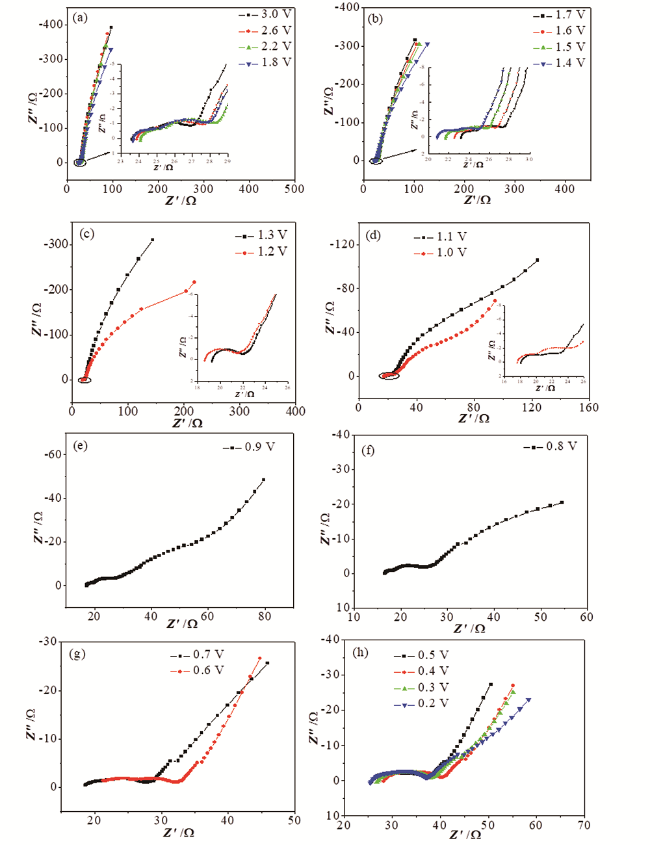
为此,我们研究了电极片厚度的不均性对锂离子电池电极EIS谱特征的影响。实验中我们在涂制电极片的过程中,特意制备出厚度有一定差异的石墨电极片(以下简称“厚度非均匀石墨电极”),并运用EIS研究这一石墨电极的首次充放电过程,实验结果如图7所示[64]。研究发现,当电极极化电极电位低于0.6 V时,EIS的一个重要特征是中频区域半圆开始逐渐分裂为两个相互独立的半圆。至电极极化电位为0.5 V时,EIS由完全相互分离的四个部分组成,即高频区域半圆、中高频区域半圆、中低频区域半圆和低频区域的斜线。等效电路拟合结果分析表明,中高频区域半圆和中低频区域半圆都与电荷传递的过程有关,表明电极片厚度的不均性是造成中频区域半圆0.6 V以下分裂的原因。
为了深入理解上述现象,我们在图8中给出了厚度均匀和非均匀石墨电极模型示意图。这里我们只考虑两种最简单的情况,即多孔电极的厚度均匀(L)或包含两种厚度(L1和L2),而且多孔电极只包含两种颗粒尺寸大小的石墨颗粒。
对于厚度均一的石墨电极,我们通过式(41)~(44)联立,直接求解电极的总阻抗。
对于厚度非均匀的石墨电极,分别对厚度为L1和L2组成的部分,根据式(41)~(44)联立,我们可以先求出厚度分别为L1和L2的两个多孔电极的阻抗值和,则包含两种厚度(L1和L2)的非均匀石墨电极的总阻抗可表示为:
其中θL1为厚度为L1的电极占整个电极的百分比。
根据上述对厚度均匀和非均匀石墨电极阻抗的求解方法,我们通过计算机数值模拟,探讨了混合颗粒比例、分层的厚度差异等因素对厚度均匀和非均匀石墨电极阻抗谱特征的影响[65]。研究发现,影响电极阻抗谱的中频区域半圆发生分裂情况的主要有两个因素,分别为:(1)电极中颗粒粒径的分布情况;(2)非均匀电极的厚度分布情况。也就是说,电极的阻抗谱特征中出现中频区域半圆发生分裂情况的前提条件就是电极中至少要包含两种不同粒径的颗粒,而电极片的厚度不均匀则能够有效地促进电极阻抗谱特征中出现中频区域半圆发生分裂的情况。
2.3.2 两相多孔电极的分散孔模型
两相多孔电极的分散孔模型,也称为圆柱孔模型(Cylindrical pore model)或均匀传输线模型(Transmission line model),是主要用于描述多孔电极阻抗特征的理论。该模型假定多孔电极是由彼此不相交联的许多单孔的组合,因而宏观电流是所有单孔中电流的总和。分散孔模型最简单的情形如图9所示,模型包含一个长度为l、半径为r充满电解液的圆柱孔。假定只有孔壁是可导的,并表现为理想极化电极的行为。圆柱孔内不存在直流电流,交流电流通过电解液流入孔内对孔壁上的双电层进行充电。
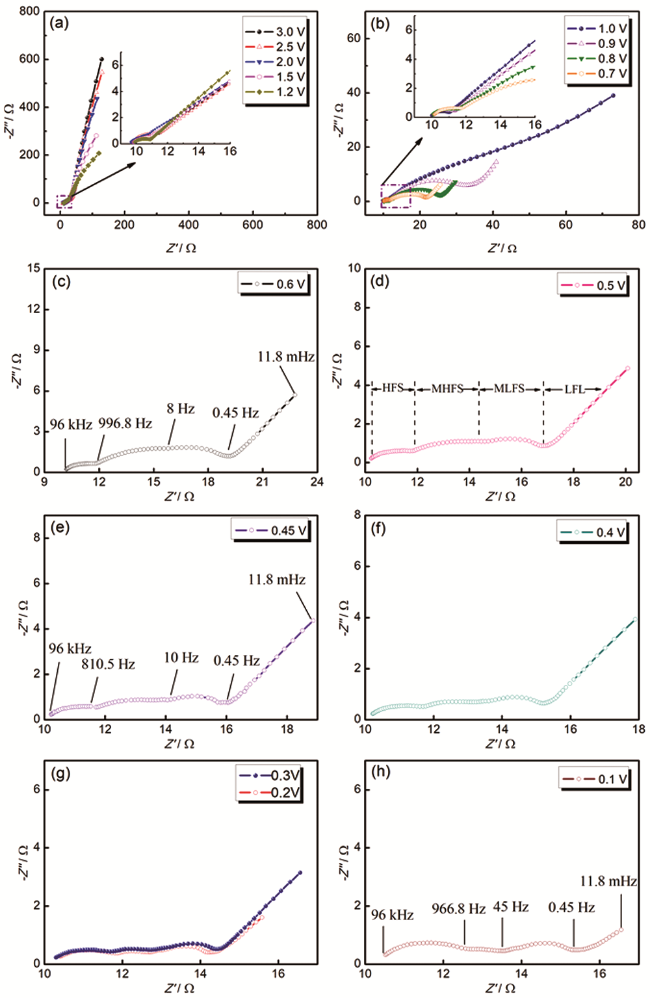

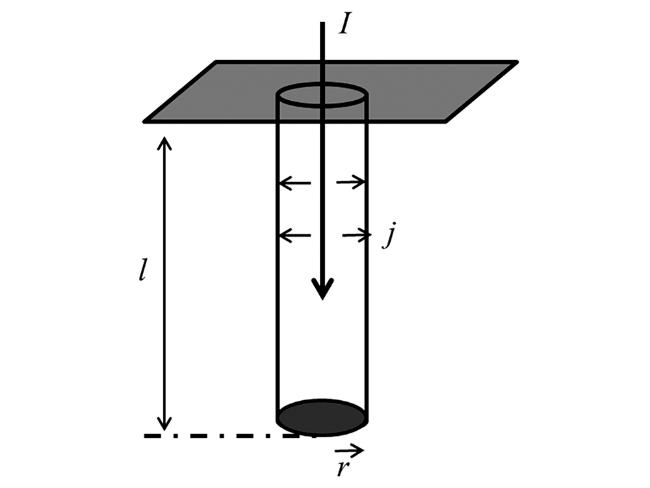
交流电流I进入孔内,并流向孔壁。由于电解液中欧姆电压降(电阻电压降),在孔内随着穿透深度的增加,流向孔壁的电流密度j不断减小。de Levie首先提出了基于圆柱孔模型的多孔电极阻抗的计算公式,迄今一直是处理此类问题的基础[66]。为便于进行数学处理,根据上面的描述,可做出以下假设:(1)多孔电极内的孔是圆柱形的。(2)只有孔壁是可导的,电极材料的电阻为零(电极材料的电导率为无穷大,电阻率ρe=0)。(3)圆柱孔内充满着电阻率为ρs(Ω·cm)的支持电解质。(4)交流电压降只存在轴线的方向,即不存在放射状的电压降。
因此,孔壁的单位面积阻抗(比阻抗)可表示为(单位为Ω·cm2),其中Cdl为单位面积电容(比电容,单位为F·cm-2)。交流电压矢量,,进入孔内后,由于电解液中的欧姆电压降将从其在孔口处的初始值随着孔深的增加逐渐降低。流向孔壁的交流充电电流矢量,同样将从其在孔口处的初始值随着孔深的增加逐渐降低。因此,上述问题可以通过两个分别描述沿孔隙的电流和电压降的公式表示:
其中参数x的变化范围为从在孔口处为0到孔底处为l之间,为单位孔长度的阻抗(Ω·cm)。对理想极化电极而言,其中是孔壁的总阻抗,是单位孔长度的双电层电容。也可以写为为单位面积双电层电容(F·cm-2),是孔壁总表面积,是在孔中单位孔长度的电解液电阻,(Ω·cm-1),是充满电解液的孔的总电阻,(Ω)。式(48)中对x进行二次微分,可得:
以及如下边界条件:
式(49)的解为:
式(51)指示,交流信号的振幅进入多孔电极的圆柱孔后,随在孔内的距离x增加而减小。
为了求得总阻抗,必须计算电位和电流矢量在孔口处的比值。在孔口处的电位梯度可表示为
因而单个圆柱孔的阻抗可表示为:
式(53)也可改写下面更为有用的形式:
其中:
是多孔电极的无量纲导纳。当孔外的电解液电阻为Rsol,对n个相同的孔而言,总阻抗为:
单个圆柱孔阻抗的Nyquist图如图10所示,巨大的阻抗值是因为单个圆柱孔的表面积非常小,其阻抗谱特征主要包括两个部分,即高频区域为一与实轴呈45°的直线和低频区域的一条垂线。在极高频区域Λ的值非常大,→1,式(54)可以简化为
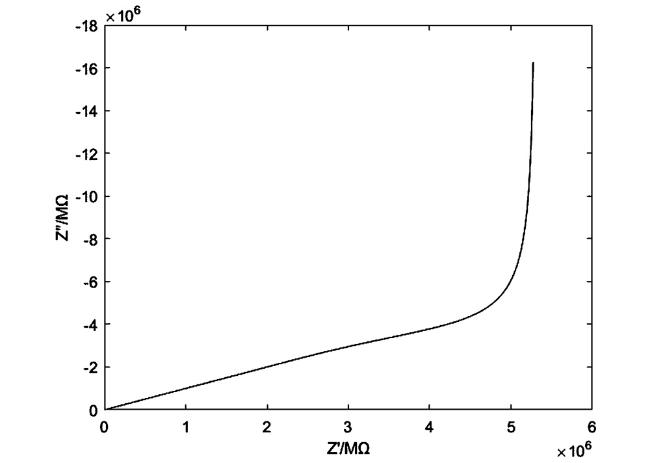
考虑到式(57)指示单个圆柱孔阻抗在高频下实部与虚部是相同的,因此在Nyquist图中呈现为与实轴呈45°的直线。
在低频区域,式(54)可表示为:
对应于的串联连接,因此在Nyquist图中呈现为电阻为的电容性直线,其电容值等于孔壁的总电容值。
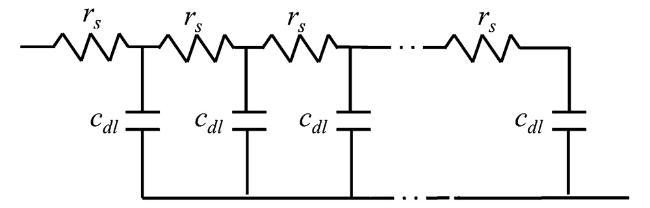
图11 均匀传输线模型示意图均匀传输线表示理想极化多孔电极的阻抗;rs和cdl分别是孔长的一个小元素的溶液电阻和双层电容Fig. 11 Schematic diagram of uniform transmission line model. Uniform transmission line representing impedance of flooded ideally polarized porous electrode; rs and cdl are the solution resistance and double-layer capacitance, respectively, of a small element of the pore length |
圆柱孔模型是真实多孔电极的一个理想化近似模型。Keiser等[69]研究了具有不同孔形的多孔电极的阻抗特征。他们认为,孔形的变化主要影响高频区域阻抗谱特征,孔形为圆柱孔时高频区域与实轴近似呈45°的直线,当孔形为梨形时高频区域演变为一个半圆。阻抗谱高频区域谱特征的这些变化是几何效应造成的,与孔中双电层电容和电解液电阻的综合作用有关。由于在低频区域所有的电极都是理想极化电极,交流信号都能完全贯穿整个孔,低频区域都呈现为一条垂线,不受孔形的影响。
其中rs和re是电解液和电极材料单位孔长度的电阻(Ω·cm-1),λ为交流信号的穿透深度。
其中为每单位孔长度的电极界面阻抗(单位Ω·cm)。
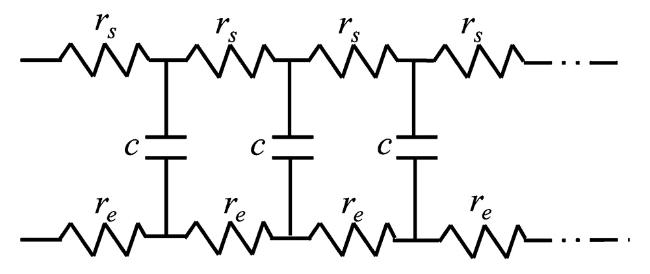
以上都是基于理想极化电极的分析结果,即假定孔内不存在法拉第过程。当孔内存在法拉第反应时,情况会变得比较复杂一些,其中最简单的一种情况就是假定孔内不存在直流电流,也就是在孔内不存在直流电压降和浓度差,系统处于平衡状态。因此,法拉第阻抗沿孔内为一常数。然而,孔内将存在交流电流和交流电压降。对于这一种情况,在假定电极材料为良导体(电阻率为0)的情况下,对圆柱孔的阻抗进行了求解。求解的过程仍然是基于求解式(47)和(48),与不存在法拉第过程的圆柱孔的阻抗求解过程相比,只是假定单位孔长度阻抗中包含表征法拉第反应的电荷传递电阻,即:
其中rct和cdl分别为单位孔长度的电荷传递电阻和孔壁电容,Rct和Cdl分别是单位面积电荷传递电阻和孔壁电容。求解的结果,与式(53)和(54)完全一样,只是孔壁阻抗变为电荷传递电阻和双电层电容并联求和的结果:
同时参数Λ重新定义为:
基于上述二相多孔电极的分散孔模型理论,Ogihara等[75,76]发展一种使用对称电池进行阻抗测试的分析技术(EIS-SC),在EIS-SC技术中,对称电池中使用两个完全相同并且测试以前具有相同电极极化电位的极片作为正负极,这样就避免了因为对电极的干扰而无法对数据进行解析的问题。实验中使用层压式软包电池,当测定多孔电极的电子电阻时,EIS测试中将两个完全相同电极片直接接触组装成电池(例如正极-正极模式),中间不放置隔膜。当测定多孔电极的孔内离子电阻时,EIS测试中在两个完全相同的电极片中间放置隔膜组装成电池(例如正极-隔膜-正极模式)。当测定多孔电极的电荷传递电阻时,在EIS测试之前,先将待测电极与对电极(如果待测电极为正极,则对电极为负极)进行充放循环后,预充至某个固定电极极化电位(一般对应于50% SOC),之后取出极片组装成对称电池,进行EIS测试。通过上述方法,能够实现对多孔电极的一些重要表征参数实现测定,并可探讨它们随温度/极片厚度的变化规律。
Malifarge等[44]则建立了在对称扣式电池中(以石墨作为电极活性材料),通过运用均匀传输线模型分析电极在阻滞条件下(不存在法拉第过程)的电极阻抗谱,获得多孔电极孔曲折率的方法。
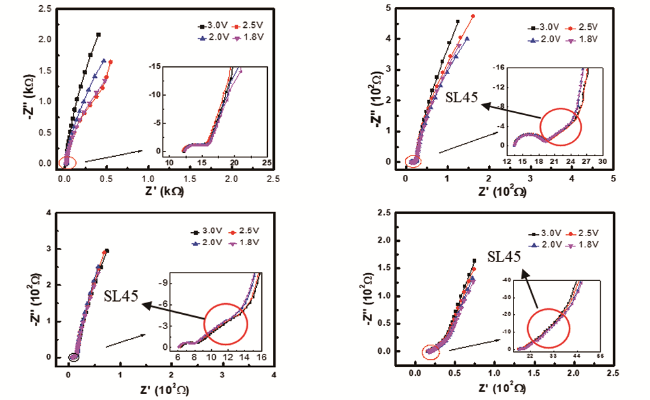
我们认为,既然在对称电池中获得的EIS中存在与多孔电极特性相关的谱特征,同时与多孔电极特性相关的谱特征是多孔电极的本质特征,理论上应该不会因为EIS测试体系(电解池)的不同而改变,因此在常规的三电极电解池体系中获得的EIS中应该同样包含与多孔电极特性相关的谱特征。为此,我们运用EIS测试了开路电位下不同厚度石墨电极片的EIS谱特征,其结果如图13所示[77]。可以看出,厚度0.2 mm与0.1 mm的阻抗谱特征相比,在阻抗谱的中频区域即高频区域半圆与低频区域一段圆弧之间出现了一条与实轴近似呈45°的直线(a straight line at 45°,缩写为SL45),随着极片厚度的增加,中频区域与实轴近似呈45°的直线的长度增大,如果不考虑高频区域的半圆,只看阻抗谱的中频区域和低频区域,极片厚度为0.6 mm时的阻抗谱特征已与式(53)指示的阻抗谱特征非常相似。上述结果表明,在三电极研究体系中也能够测试到与多孔电极特性相关的谱特征。此外,我们还运用EIS测试了相对较厚(约0.15 mm)的 LiNi1/3Co1/3Mn1/3O2 电极的首次充电过程,如图14所示。发现在其阻抗谱中同样存在与多孔电极特性相关的谱特征,进一步证实在三电极研究体系中能够观察到与
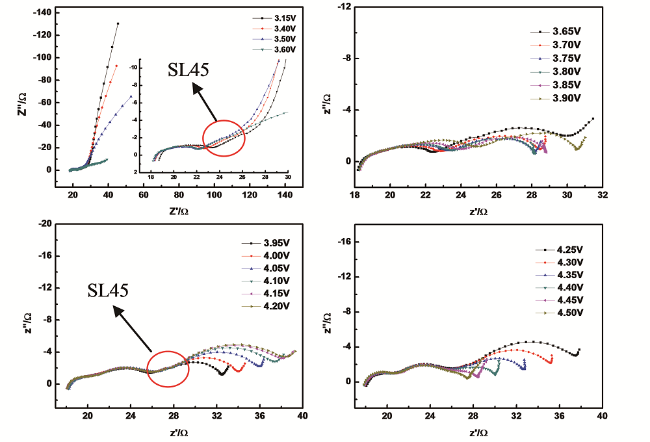
多孔电极特性相关的谱特征。可以合理预测,分析与多孔电极特性相关的谱特征在首次充放电过程以及长期充放电循环过程中,EIS谱特征中与多孔电极特性相关的谱特征等效电路拟合所获得的表征参数随电极极化电位和充放电循环周数的变化,对于深入理解锂离子电池的容量和功率衰减机制将有重要作用,对于发展基于厚电极的高比能电池将起到关键性的指导作用。
3 锂离子电池电极的电化学阻抗谱特征
3.1 锂离子电池负极的EIS谱特征
3.1.1 石墨电极的EIS谱特征
通常锂离子嵌入石墨电极过程EIS的Nyquist图由三部分组成:高频区域和中频区域各存在一个半圆,低频区域则为一条斜线。根据Aburch等[11]的观点,高频区域半圆应归属于锂离子通过石墨电极表面SEI膜的扩散迁移、中频区域半圆与电荷传递过程相关,而低频区域的斜线则反映了锂离子在石墨电极中的固态扩散过程。虽然人们对中频区域半圆和低频区域斜线的归属意见比较一致,但对高频区域半圆的归属一直存在争议,这是因为如果将高频区域半圆归属于锂离子通过石墨电极表面SEI膜的扩散过程,那么必然会得到两个推论:(1)石墨电极表面通常存在初始的SEI膜,在开路电位(Open circuit potential, OCP)下,也不会与电解液组分发生反应形成SEI膜,因此,在经历电化学循环扫描前和OCP下,石墨电极EIS的Nyquist图中应该不存在高频区域半圆,即此时石墨电极表面不存在SEI膜;(2)SEI膜主要在首次充放电过程中形成,因此石墨电极在首次阴极极化过程中,应该能够观察到高频区域半圆的出现和成长过程。然而,一些研究者发现[78,79,80],在经历电化学循环扫描前和OCP下,石墨电极EIS的Nyquist图中就存在高频区域半圆,且在石墨电极首次阴极极化过程中,EIS的高频区域几乎不发生任何变化。因此,他们认为高频区域半圆应主要归因于接触阻抗问题。
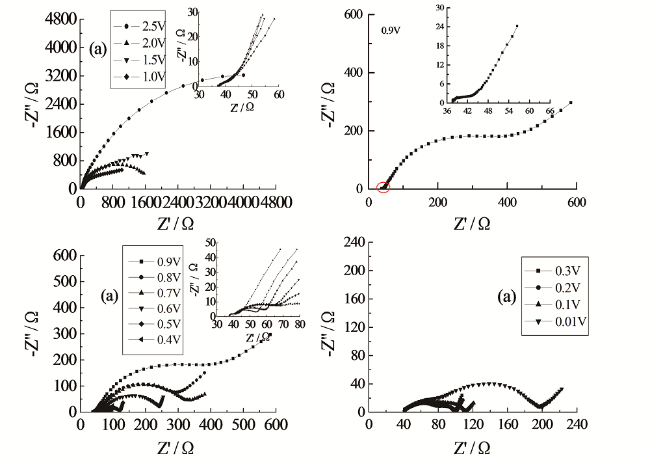
我们认为,如果高频区域半圆与接触阻抗问题有关,那么通过适当的方法应该能够减小甚至消除接触阻抗,进而消除在经历电化学循环扫描前和OCP下石墨电极EIS中存在的高频区域半圆。为此,在电极片的制备过程中,我们通过对集流体表面粗糙化(增大电极膜和集流体之间的结合力与接触面积)、适当增加黏合剂(PVDF-HFP)在溶剂(N-甲基吡咯烷酮)中的溶解时间和搅拌强度(确保黏合剂完全均匀溶解)、延长电极浆料的搅拌时间和强度(搅拌的同时进行超声震荡)、增加电极片的辊压强度(60 MPa)、增加电极膜的厚度(0.15 mm左右)来降低接触阻抗。实验中获得的石墨电极(石墨碳纤维,本文如无特别指明,电极材料一般均指商品化电极材料,研究体系为三电极玻璃电解池,EIS测试频率范围为105~10-2 Hz)在首次阴极极化过程中EIS的Nyquist图随电极极化电位的变化如图15所示[81,82]。与预期结果相一致,研究发现在经历电化学循环扫描前和OCP下,石墨电极的Nyquist图在整个测试频率范围内为一段圆弧,不存在高频区域半圆。在SEI膜的成膜电位0.9 V时[83],高频区域出现了一个小半圆。此时,Nyquist图由三个相互分离的部分组成,即高频区域和中频区域各存在一个半圆,低频区域则为一条斜线。上述结果与将高频区域半圆归因于锂离子通过石墨电极表面SEI膜的扩散过程所得到的推论结果是完全一致的,表明石墨电极EIS的高频区域半圆不仅与锂离子通过石墨电极表面SEI膜的扩散过程有关还与接触阻抗有关,只有将接触阻抗基本消除或减小到一定程度,才能观察到因SEI膜的形成而导致的EIS谱特征的变化。
然而,图15实验结果的获得具有一定的偶然性,这主要是因为要消除接触阻抗所需优化控制的实验条件太多,通常情况下很难获得接触阻抗基本被消除的电极片。我们进一步研究发现,只要将接触阻抗减小到一定程度,虽然在开路电位下,EIS谱中就存在高频区域半圆以及由此观察不到高频区域半圆的出现过程,但通过等效电路的拟合结果,仍然可以很好观察到因SEI膜的形成过程,造成高频区域半圆的变化(即高频区域半圆对应的阻抗值变化)。因此,运用EIS研究石墨电极首次阴极极化过程,能够直接研究SEI膜的形成机制及其影响因素,并将这一方法应用于研究电解液中杂质[84,85]和添加剂[86,87,88]对SEI膜形成机制的影响,获得了较好的研究结果。
3.1.2 硅电极的EIS谱特征
硅具有高达4200 mAh/g 的理论比容量,在电化学储锂过程中, 平均每个硅原子结合4.4个锂原子得到Li22Si5合金相,材料的体积变化达到 300%以上[89],在电极内部产生较大的机械应力,这一方面易造成电极活性材料的破碎、粉化和电极活性物质与集流体之间逐渐脱开,致使电极材料活性颗粒与集流体失去良好的电接触和机械接触;另一方面易造成电极表面SEI膜的破裂,在新裸露出的硅颗粒表面不断形成新的SEI 膜,导致充放电效率降低,容量衰减加剧。
可以合理预测,增强复合电极的导电性,抑制充放电过程中活性材料颗粒与锂反应时因体积膨胀而导致的电极破碎、粉化,保持材料颗粒良好的电接触、机械接触不仅是改善硅基材料电化学性能的重要方法[90],也是获得硅电极EIS谱特征的基础,否则测试到的结果中可能只能观察到与电极体积剧烈膨胀的信息,而无法观察到与电化学反应相关的信息。
3.1.3 过渡金属氧化物电极的EIS谱特征
3d过渡金属由于其有价电子依次填充在次外层的d轨道上,同时d壳层没有填充满电子,电子逐个填入它们的3d轨道,从而其性质与其他元素有明显差别。它们的氧化物属于强关联电子体系,也就是说体系中存在电子和电子之间的库仑相互作用,导致3d过渡金属氧化物大多数导电性都较差,而二元3d过渡金属氧化物中的一半几乎都是Mott绝缘体。因此,3d过渡金属氧化物大多电化学反应活性不强,存在严重的电压滞后现象,导致较低的能量效率,必须减小活性物质颗粒的尺寸(一般至纳米尺度范围)。此外,锂离子与3d过渡金属氧化物发生转化反应过程中,一方面会生成过渡金属纳米颗粒,增进电极的导电性;另一方面会生成大量的低密度的氧化锂使得电极体积剧烈膨胀,应力的累积可能导致活性物质的粉化并且从集流体上脱落,以及电极表面SEI膜的破裂,最终导致容量衰减。
与硅电极类似,将3d过渡金属氧化物与碳材料复合制备成3d过渡金属氧化物/C复合材料,同时适当提高复合电极中导电剂和黏合剂的比例,增进电极的导电性能,缓冲3d过渡金属氧化物与锂离子发生转化反应时体积的变化,增加复合材料的结构稳定性,是获得3d过渡金属氧化物电极EIS谱特征的基础,否则测试到的结果中可能只能观察到与电极体积剧烈膨胀的信息,而无法观察到与转化反应相关的信息。在限制了转化反应发生过程中电极活性材料体积产生巨大变化的前提下,影响到3d过渡金属氧化物电极EIS谱特征的因素主要有4项:接触阻抗、锂离子通过SEI膜的扩散迁移、活性材料的电子电导率和电荷传递过程。
20世纪80年代,α-Fe2O3就曾经被作为锂离子电池负极材料研究过[92,93,94],它作为锂离子电池电极材料具有高达1007 mAh/g的理论比容量,而且具有相对较好的导电性以及廉价、无毒等优点受到较多的关注,是基于转化反应机制储锂材料中3d过渡金属氧化物的典型代表。早期文献曾经认为α-Fe2O3是嵌合物电极材料[95],Thackeray等[93]研究发现少量的锂离子与α-Fe2O3之间会发生嵌入反应,生成LixFe2O3(0≤x≤2),而过量锂离子则进一步与LixFe2O3发生转化反应生成Li2O和Fe相,因而锂离子与α-Fe2O3的反应为连续的锂离子嵌入反应、转化反应两步机制,可表示为:
如前文所述,将α-Fe2O3与碳材料复合制备成α-Fe2O3/C复合材料不仅是改善其电化学性能的有效手段[96,97,98],也是获得其典型EIS谱特征的基础。因此,我们采用高温固相反应法制备了α-Fe2O3/C复合材料,并运用EIS研究了α-Fe2O3/C复合材料电极首次放电过程中EIS谱特征随电极极化电位的变化,其结果如图17所示[99]。研究发现,在锂离子与α-Fe2O3发生电化学反应过程中,α-Fe2O3/C复合材料电极的EIS谱特征由四个明显相互分离的部分组成,即高频区域、中频区域和低频区域各存在一个半圆和更低频区域的一条斜线。等效电路拟合分析结果表明,高频区域半圆应主要归属于锂离子通过SEI膜的扩散过程和接触阻抗问题,中频区域半圆是与α-Fe2O3/C复合材料的电子电导率相关的半圆,而低频区域半圆和更低频区域的斜线则分别归属于与电荷传递过程和锂离子活性材料颗粒内的固态扩散过程。
3.2 锂离子电池正极的电化学阻抗谱特征
锂离子电池正极活性材料的电化学以及物理性质与材料的晶体结构以及电子结构紧密关联,特别是材料的电子结构。按导电机制不同,锂离子电池正极活性材料可分为n型半导体电极、p型半导体电极和绝缘体电极。锂离子电池电极活性材料的导电机制对锂离子电池正极的电化学阻抗谱特征有重要影响。
3.2.1 p型半导体电极的EIS谱特征
LiCoO2为典型的p型半导体(带隙宽度Eg =2.7 eV)[106,107,108],主要依靠空穴导电。已有的研究结果表明[109,110,111,112],LiCoO2在锂离子脱出过程中,存在导电性能快速增加的过程,即材料的导电性质由近乎不导电的绝缘体(或半导体)转变为金属导体的物理转变,这一转变过程一般称为“金属-绝缘体”转变(Metal-insulator transitions,也称为Mott转变)。也就是说,LixCoO2在未发生锂离子脱出时(即x=1时),表现出近乎不导电的绝缘体导电性质;在锂离子脱出初期阶段,即1>x> 0.95时,LixCoO2材料的电子电导率缓慢增大;在锂离子脱出中期阶段,即0.95>x>0.75 时,LixCoO2材料的电子电导率快速增大;当x<0.75 时,LiCoO2材料表现为金属的导电性质。
我们认为,欲获得LiCoO2电极的EIS谱特征中存在与活性材料在锂离子脱出嵌入过程中电子电导率变化相关的半圆,必须首先要保证电极中的接触阻抗较小(因为接触阻抗和电子在活性材料本体中的输运,本质上都属于电子输运过程的一部分,有可能在EIS谱中互相重叠),同时应该保证电极片中导电剂形成了较好的导电网络,使每一个活性材料颗粒都与导电剂颗粒形成良好的接触。根据这一假设,我们对电极片的制备过程按前述方法进行了优化,同时增加了电极中导电剂和黏合剂的含量(质量百分比均为10%),实验结果如图19所示[123]。研究发现,与文献中已报道的结果不同,LiCoO2电极在OCP下(3.6 V)的EIS谱中包含一个压扁拉长的高频区域半圆、一个巨大的中频区域半圆及低频区域的一条斜线。当电极极化电位高于4.15 V时,Nyquist图中清晰地体现出三个半圆及一条斜线的谱图特征。等效电路分析结果证实,中频区域半圆与电极活性材料的电子电导率相关。
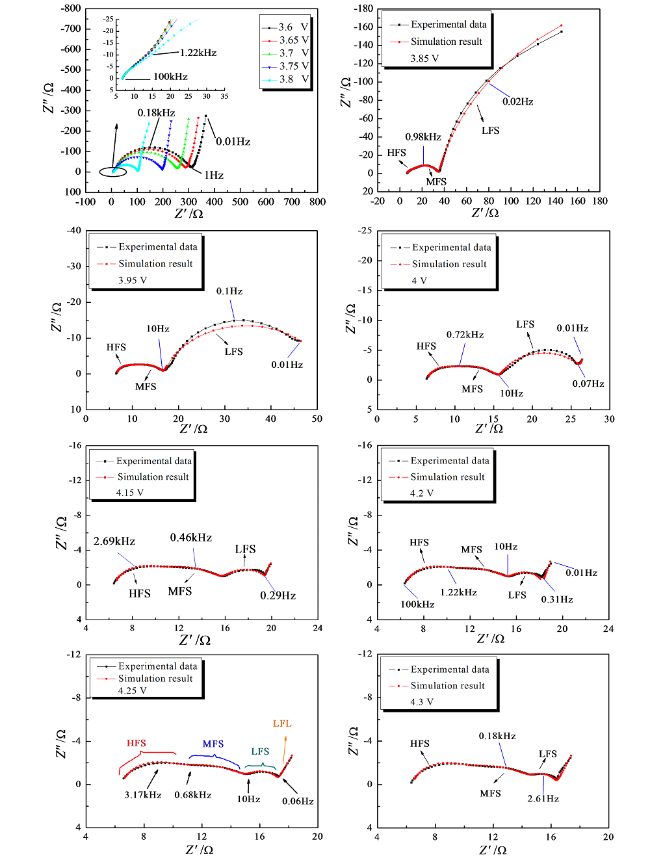
LiNi1/3Co1/3Mn1/3O2的晶体结构与LiCoO2的晶体结构非常相似,都具有α-NaFeO2型结构,同样为p型半导体。我们进一步运用EIS研究了LiNi1/3Co1/3Mn1/3O2电极的首次充放电过程,发现LiNi1/3Co1/3Mn1/3O2电极与LiCoO2电极具有相似的阻抗谱特征[124]。可以推测,所有具有α-NaFeO2型结构的p型半导体电极都具有相似的阻抗谱特征,EIS的Nyquist图中包含四个部分:与锂离子通过电极活性材料表面SEI膜扩散过程有关的高频区域半圆,与电极活性材料电子输运性质相关的中频区域半圆,与电荷传递过程相关的低频区域半圆以及极低频区域与固态扩散相关的斜线。
3.2.2 n型半导体电极的EIS谱特征
与p型半导体不同,n型半导体主要依靠自由电子导电。根据Mott给出的关于电导率的一般公式[125],n型半导体材料本体电子电导率σ可表示为:
其中β为描述波函数分布的参数,r为两个临近的不同价态的过渡金属离子位置之间的距离,ve为电子跳跃频率,cx0为低价态过渡金属离子的浓度(载流子-电子的浓度,),c(1-x0)为高价态过渡金属离子的浓度(电子跃迁的空穴浓度),k为Boltzmann常数,T为热力学温度。
3.2.2.1 尖晶石LiMn2O4电极的EIS谱特征
尖晶石LiMn2O4为n型半导体,其电子的传导主要依靠电子在低价(Mn3+)和高价(Mn4+)离子之间的跃迁实现[126,127,128]。从式(66)可以得出,LiMn2O4材料的电子电导率主要由两个方面的因素决定:(1)载流子(Mn3+中的电子)和空穴(Mn4+中的空穴)的浓度,即x0的值;(2)载流子的跃迁长度(Mn3+-Mn4+原子间的距离),即r的值。对尖晶石LiMn2O4材料而言,在锂离子脱出之前,即开路电位下,x0=1/2。在锂离子脱出过程中,伴随着Mn3+氧化为Mn4+导致x0不断减小,将导致尖晶石LiMn2O4电子电导率降低(即x0(1-x0)的值减小);同时,锂离子的脱出会引起尖晶石结构中Mn3+-Mn4+原子间距离的收缩,即r减小,将导致尖晶石LiMn2O4的电子电导率增高。由上述讨论可知,在充放电过程中,尖晶石LiMn2O4材料本体电子电导率会随着充放电过程的进行而发生变化,其随电极极化电位或SOC变化的规律是上述两个方面因素相互作用的结果。因此,尖晶石LiMn2O4电极的EIS谱特征应该与LiCoO2电极的EIS谱特征类似,即Nyquist图中存在与活性材料电子电导率相关的一个半圆。不同之处在于,二者导电机制不同,电子电导率随电极极化电位(或SOC)的变化规律不同。
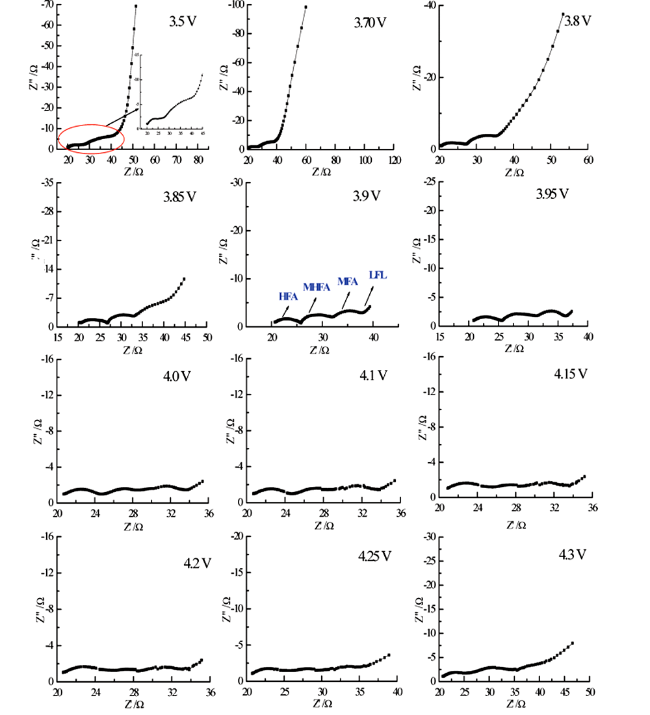
我们运用EIS研究了10 ℃下尖晶石LiMn2O4电极的首次充放电过程,其充电过程实验结果如图20所示[129]。研究发现,在OCP下(3.5 V),尖晶石LiMn2O4电极的EIS谱特征由两个半圆与一条斜线组成。当电极极化电位升高至3.85 V以上时,EIS谱特征包括四个部分:高频区域、中频区域和低频区域各存在一个半圆,以及更低频区域与扩散相关的斜线。等效电路拟合分析结果表明,高频区域半圆与锂离子扩散迁移通过SEI膜的过程有关,中频区域半圆与尖晶石LiMn2O4电极活性材料的电子电导率有关,低频区域半圆与电荷传递过程有关,而更低频区域的斜线则反映了锂离子在尖晶石LiMn2O4电极材料中的固态扩散过程。上述研究结果与理论分析结果相一致,即尖晶石LiMn2O4电极的EIS谱特征中同样存在与活性材料电子电导率相关的一个半圆。
3.2.2.2 LiFePO4电极的EIS谱特征
历史上发表过很多关于LiFePO4电极EIS研究的论文,然而对于LiFePO4电极的EIS谱特征的解释一直存在一些争论。一些研究者实验中获得的LiFePO4电极的EIS谱特征由两部分组成[133,134,135,136,137,138],即高频区域的一个半圆和低频区域的一条斜线,并将它们分别归属于电荷传递过程和锂离子在电极活性材料颗粒中的固态扩散过程。然而Jamnik等[139]认为,LiFePO4电极的EIS谱特征中高频区域半圆应该归属于电极材料和集流体之间形成的接触阻抗。此外,Gaberscek等[140]报道,LiFePO4电极的EIS谱特征由三个部分组成,即分别在高频区域、中频区域存在一个半圆和低频区域的一条斜线。他们将高频区域半圆归属于电极材料和集流体之间形成的接触阻抗,但无法确定中频区域半圆是应该归属于电极活性材料颗粒之间的接触阻抗还是电荷传递过程。
LiFePO4为n型半导体,理论上讲,应该具有与尖晶石LiMn2O4电极相似的阻抗谱特征,但LiFePO4的导电性能很差,近乎绝缘,因此其与活性材料电子电导率相关时间常数的特征频率有可能会超出仪器通常的频率测试范围(105 ~ 10-2 Hz),而观察不到。同时,LiFePO4非常稳定,其表面形成的SEI膜可能会非常薄(抑或是不存在),与锂离子通过SEI膜扩散迁移过程相关的时间常数也有可能会超出仪器通常的频率测试范围或超出仪器测试精度,而观察不到。为克服LiFePO4导电性能差的障碍,一般需要对LiFePO4活性材料颗粒进行表面碳包覆和减少LiFePO4活性材料颗粒的尺寸,以改善其电化学性能。考虑到LiFePO4活性材料颗粒的粒径很小(一次颗粒的粒径一般在几十纳米尺度范围),电极制备过程中活性材料颗粒分散较为困难,尤其是在实验室条件下,当导电剂的量较小时,很难保证LiFePO4活性材料颗粒与导电剂颗粒之间实现微观上的均匀混合,即每一个LiFePO4活性材料颗粒都与导电剂颗粒接触良好。由此推测,上述文献中报道的LiFePO4电极的EIS谱特征呈现出很大不同,与电极片的制备过程不同有关。
基于以上考虑,我们研究了导电剂的含量对LiFePO4电极的EIS谱特征的影响,实验中我们选用石墨作为导电剂,以避免使用导电炭黑带来的分散困难和导电炭黑量大时电极片粘合力下降的问题[141]。研究发现,当导电剂含量低于30%时,LiFePO4电极EIS谱特征都由高频区的一个半圆和低频区的一条斜线两部分组成。在电极极化电位测试范围内,LiFePO4电极的EIS谱特征随电极极化电位的升高,几乎没有发生任何的变化。在相同的电极极化电位下,随着导电剂含量的增加,高频区域半圆逐渐变小,表明导电剂的含量对LiFePO4电极的界面阻抗有很大的影响。等效电路拟合结果分析显示,高频区域半圆不能归属于电荷传递过程而只能归属于接触阻抗问题。
当导电剂含量增加到50%时,LiFePO4电极的EIS谱特征与导电剂含量低于30%时完全不同,实验结果如图21所示在锂离子脱出阶段,始终由三个部分组成,即高频区域和中频区域各存在一个半圆以及低频区域的一条与固态扩散相关的斜线。等效电路模拟分析结果表明,中频区域半圆应归属于电荷传递过程。
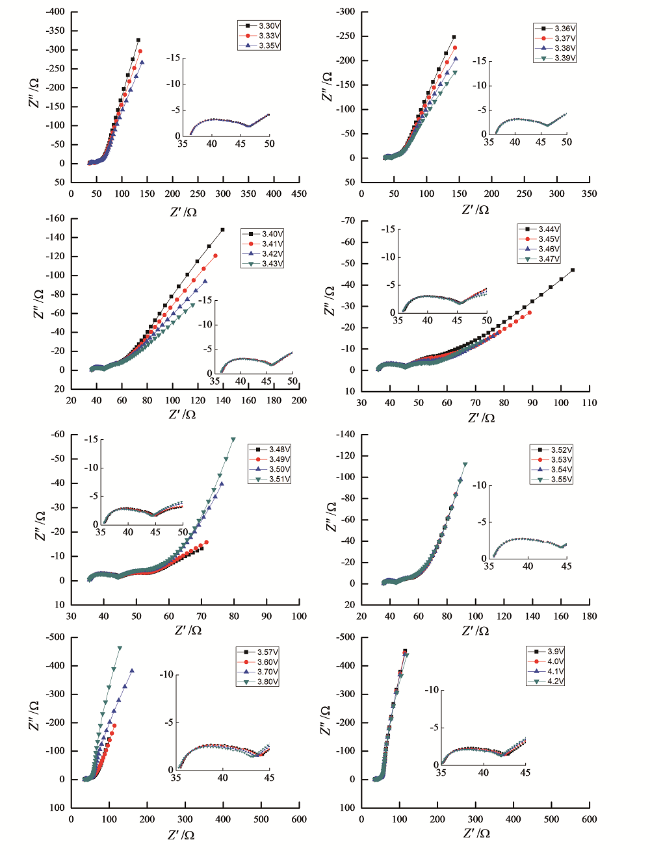
上述结果表明,由于LiFePO4活性材料颗粒粒度较小,造成电极片制备过程中活性材料与导电剂实现均匀分散困难,进而导致接触阻抗较大,因此导电剂的含量对LiFePO4电极的EIS谱特征影响很大。当LiFePO4电极的制备过程经过优化后,其典型的EIS谱特征由三个部分组成,即主要与接触阻抗有关的高频区域半圆,主要与电荷传递过程有关的中频区域半圆和反映锂离子在电极活性材料颗粒中固态扩散迁移的低频区域斜线。
3.2.2.3 尖晶石Li4Ti5O12(LTO)电极的EIS谱特征
尖晶石Li4Ti5O12一般在锂离子电池中用作负极材料,但其晶体结构以及电子结构都与尖晶石 LiMn2O4类似,只是锂离子在其中的嵌入脱出电极电位相对较低。因而,本文中在探讨其EIS谱特征时,我们将其作为n型半导体正极材料进行讨论。
同理,尖晶石Li4Ti5O12中电子的传导是通过电子从低价钛(Ti3+)向高价钛(Ti4+)的跃迁来实现的。从式(66)可知,尖晶石Li4Ti5O12材料的电子电导率同样是主要由两个方面的因素决定:(1)载流子-电子的浓度(Ti3+提供)和空穴的浓度(Ti4+提供);(2)载流子的跃迁长度(Ti3+-Ti4+原子间的距离)。充放电时,锂离子的嵌入和脱出对钛酸锂材料的晶型结构几乎没有影响,a值仅从0.836 nm增大到0.837 nm[142],
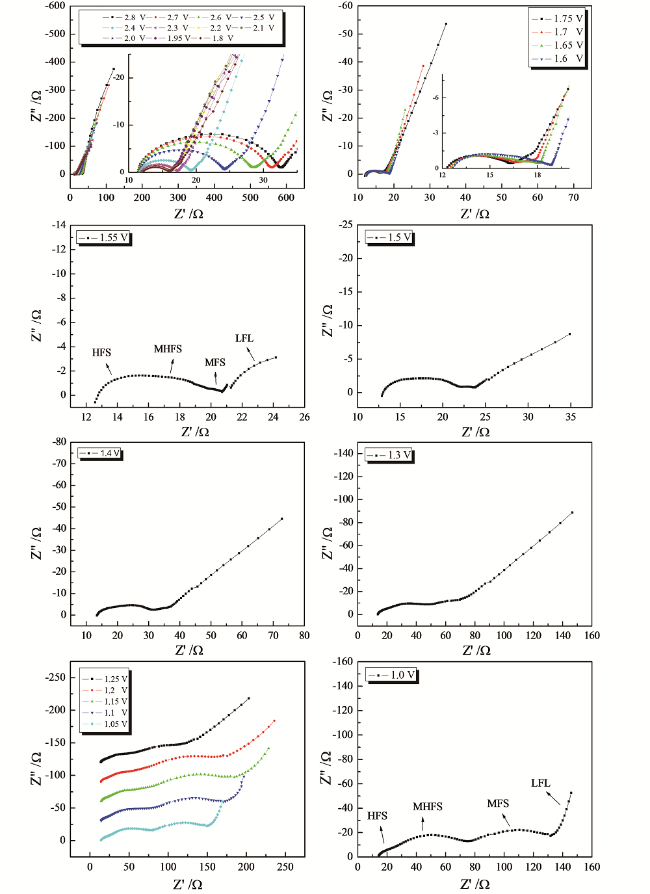
也就是说Ti3+-Ti4+原子间的距离在充放电过程中几乎不变。因此,与尖晶石LiMn2O4不同,载流子的跃迁长度对尖晶石型Li4Ti5O12材料的电子电导率几乎没有影响,影响尖晶石Li4Ti5O12材料电子电导率的主要因素是载流子(电子)浓度和空穴浓度。在锂离子嵌入尖晶石Li4Ti5O12以前,尖晶石Li4Ti5O12活性材料中四价钛占据主导地位,x0近似等于0,此时尖晶石Li4Ti5O12几乎是绝缘体。随着锂离子嵌入过程的进行,四价钛不断转化为三价钛,空穴浓度不断降低而载流子浓度不断增大,即x0不断增大,材料的电子电导率不断增大。当每个尖晶石Li4Ti5O12结构单元中嵌入2.5个锂离子时,x0=1/2,材料的电子电导率达到最大,之后随着锂离子的嵌入,材料的电子电导率会逐渐减小。
因此,尖晶石Li4Ti5O12材料电子电导率相对较低,尤其在经历充放电过程之前,完美晶体几乎是绝缘体。为克服这一问题,一般需要对尖晶石Li4Ti5O12材料进行晶粒细化和表面碳包覆。商品化尖晶石Li4Ti5O12材料一般由典型的两种不同大小晶粒组成:一种晶粒粒度在0.2 ~ 0.5 μm范围内,另一种在1 ~ 1.5 μm范围内。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3.2.3 绝缘体电极的EIS谱特征
在元素周期表中,氟的电负性最强,因此与过渡金属形成的氟化物的离子键强度非常高,相应的作为电极材料时充放电电压平台也就比较高。大多数过渡金属氟化物的破键/成键电位可达到2.0 V以上,理论上能够用作正极材料使用。目前已报道的过渡金属氟化物正极材料主要有NiF2、FeF3、CuF2、TiF3、VF3、CoF2等,过渡金属氟化物一般都具有较好的氟离子导电性。但是,由于过渡金属氟化物的强离子键特征而产生大的带隙,导致它们的电子导电性能通常都很差,几乎都是绝缘体。
此外,过渡金属氟化物发生转化反应后的产物LiF是电子和离子的不良导体,因此,过渡金属氟化物电极中不存在锂离子固态扩散的过程,离子的输运过程主要是氟离子在过渡金属氟化物/金属界面快速传输至过渡金属氟化物/电解质的界面,在界面处氟离子与锂离子反应生成LiF[147]。可以推测,过渡金属氟化物电极的EIS谱特征中,不会出现与材料电子电导率相关的半圆和反映锂离子固态扩散过程的低频区域斜线。特别是,过渡金属氟化物电极的EIS谱特征中一般不会出现反映锂离子固态扩散过程的低频区域的斜线,是区别于其他类型电极的EIS谱特征的鲜明特点。
进一步研究发现,FeF3/C复合材料[148]、FeF3(H2O、Li3FeF6、Na3Fe等均呈现与NiF2/C复合材料相似的EIS谱特征。
4 结论与展望
综上所述,可以得出锂离子在嵌合物电极活性材料中的嵌入脱出过程主要包括三个基本物理化学过程:(1)电子的输运过程,主要包括两个步骤,其一是电子从外电路传输到嵌合物电极活性材料颗粒表面,这一步表现为接触阻抗问题;其二是电子在嵌合物电极活性材料颗粒内的输运问题,这一步表现为嵌合物电极活性材料的电子电导率问题,上述两个步骤在EIS的Nyquist中均会呈现为一个半圆;(2)锂离子的输运过程,主要包括两个步骤,其一是锂离子通过电极活性材料表面SEI膜的扩散过程,这一步骤在EIS的Nyquist中会呈现为一个半圆;二是锂离子在多孔电极孔隙中电解液的扩散过程,这一步骤在EIS的Nyquist中会呈现为一条与实轴近似45°的直线;(3)电化学反应过程,主要包括两个步骤,其一为电荷传递过程,这一步骤在EIS的Nyquist中会呈现为一个半圆;其二为锂离子在电极活性材料颗粒内部的固态扩散,这一步骤在EIS的Nyquist中会呈现为一条与实轴近似45°的直线;此外,电化学反应过程还包括锂离子和电子在电极活性材料颗粒内部的积聚过程,这一步骤在EIS的Nyquist中会呈现为一条垂线,但是这一垂线往往出现在极低频区域(约5 mHz附近),因而其在EIS谱中一般观察不到;以及相变过程,这一步骤在EIS的Nyquist中会呈现为一个半圆,但锂离子在嵌入化合物的嵌入脱出过程中一般体积变化较小,体相内部物理化学性质变化不大,且一般不存在剧烈的相变过程,除了一些特殊的电极活性材料如Si外[151],一般其在EIS谱中也很难观察到。值得特别强调的是,锂离子在多孔电极孔隙中电解液的扩散过程和锂离子在电极活性材料颗粒内部的固态扩散过程均在EIS的Nyquist中呈现为一条与实轴近似呈45°的直线。但与锂离子在多孔电极孔隙中电解液的扩散过程相关的直线一般出现在高频区域(在Nyquist中出现在与电荷传递过程相关的半圆之前),而与锂离子在电极活性材料颗粒内部的固态扩散过程一般出现在低频区域(在Nyquist中出现在与电荷传递过程相关的半圆之后)。因此,EIS谱特征通常会包括至少6个时间常数,但受电极活性材料本身性质不同、电极片制备工艺(包括电极活性材料与黏合剂、导电剂的比例,电极浆料的搅拌混合、涂布、烘干、辊压过程以及电极片厚度)的影响,有的时间常数在EIS的Nyquist中可能观察不到。此外,由于一些不同的物理化学过程,具有相近的时间常数,例如与接触阻抗相关的半圆和与锂离子通过电极活性材料表面SEI膜的扩散过程的半圆,它们通常会在Nyquist中重叠成一个半圆。可以说,目前EIS在锂离子电池领域的应用中面临的严重限制,仍然是其测试结果的不确定性,主要表现为由于时间常数相近,不同的物理过程或一个复杂过程的不同步骤在电化学阻抗谱中的特征相互重合,成为一个特征,导致对电极反应相关的复合阻抗谱的解释比较困难。这一问题的解决除了进一步提高阻抗谱仪器的测试精度外,发展新的阻抗谱测试技术例如非线性阻抗谱技术[152,153],从阻抗谱的高次谐波中获得更多信息,以及将阻抗谱实验研究与阻抗谱的数值模拟相结合,充分利用两者优点,将是解决这一问题的重要手段,可能也是未来电化学阻抗谱技术在锂离子电池领域中应用的重要发展方向。
最后,经过20年的发展,EIS已经成为锂离子电池研究领域内一个重要或者说是不可或缺的分析手段,但目前EIS作为一种测试方法在锂离子电池工业化生产中直接获得应用的报道还不多[154],这方面工作亟待加强,也应该是未来的一个重要发展方向。