



Contents
1 引言
2 计算模拟方法
3 C2、C3气体分离
3.1 乙烷乙烯分离
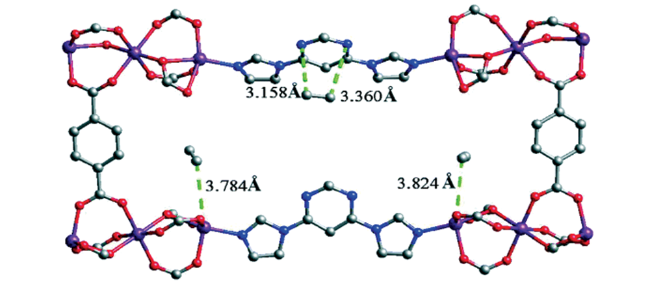
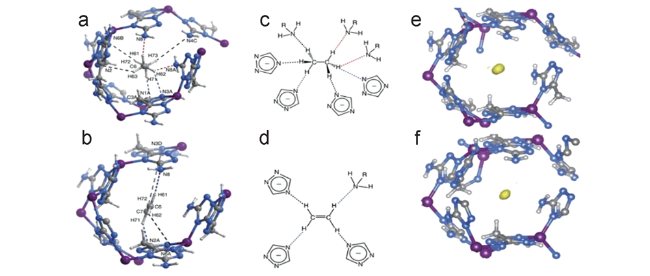
图2 计算模拟结果表明,(a) C2H6和(b) C2H4在MAF-49中的优先吸附位点。 (c) C2H6和(d) C2H4对应的主客交互的示意图。强(H…N/O<2.3 Å)、弱(2.3 Å<H…N/O<2.8 Å)和几乎可以忽略的(H…N/O<2.8 Å) C-H…N相互作用分别以红、蓝、黑虚线表示[26]Fig. 2 Preferential adsorption sites for (a) C2H6 and (b) C2H4 in MAF-49 revealed by computational simulations. Schematic representation of the corresponding host-guest interactions for (c) C2H6 and (d) C2H4. Strong (H…N/O<2.3 Å), weak (2.3Å<H…N/O<2.8 Å) and almost negligible (H…N/O<2.8 Å) C-H…N interactions are displayed as red, blue and black dashed lines, respectively[42] |
3.2 乙烯乙炔分离

图3 DFT优化后的C2H2 (a)和C2H4 (b)在TIFSIX-2-Ni-i中的吸附构型示意图,为了清晰起见,不同的网采用蓝色和绿色高亮显示。Si,黄色;F,红色;Ni,紫色[48]Fig. 3 Schematic pictures showing the DFT optimized C2H2 (a) and C2H4 (b) adsorption configurations in TIFSIX-2-Ni-i, the different nets are highlighted in blue and green color for clarity. Color code: Si, yellow; F, red; Ni, purple[48] |
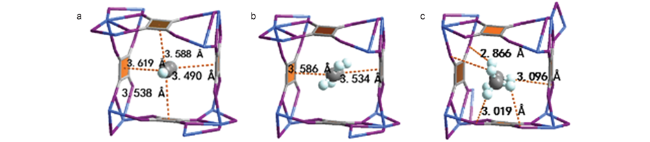
图4 DFT-D计算轻烃(C2H2, C2H4, C2H6)在Ca(环丁烯)中的吸附结合位点。(a)乙炔与Ca(环丁烯)通过π-π和“Hδ+…Oδ-”多重范德华相互作用。(b) C2H4与框架通过π-π相互作用交互距离(3.534 Å和3.586 Å)。(c) C2H6与优化结构通过“Hδ+…Oδ-”多个范德华相互作用(2.809 Å到3.613 Å)。C,灰色;Ca,蓝色;O,紫色;H,浅蓝色[49]Fig. 4 DFT-D calculated light hydrocarbons (C2H2, C2H4, C2H6) adsorption binding sites in Ca (squarate). (a) C2H2 interacts with Ca (squarate) through π-π and “Hδ+…Oδ-” multiple van der Waals interaction. (b) C2H4 interacts with the framework through π-π interactions with distances (3.534 Å and 3.586 Å). (c) C2H6 interacts with the optimized structure through “Hδ+…Oδ-” multiple van der Waals interaction (2.809 Å to 3.613 Å). C, gray; Ca, blue; O, purple; H, light blue[49] |
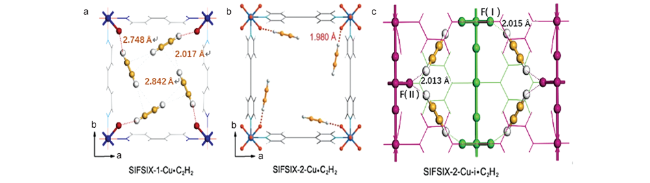
3.3 丙烷丙烯分离
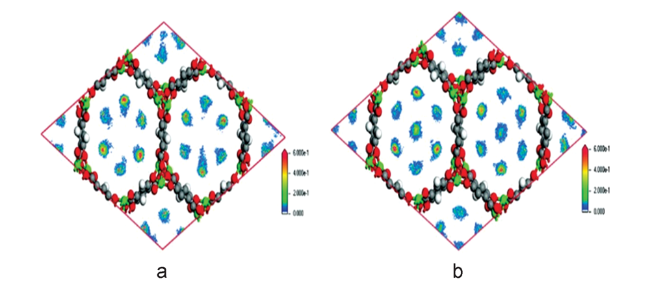
3.4 丙烯丙炔分离
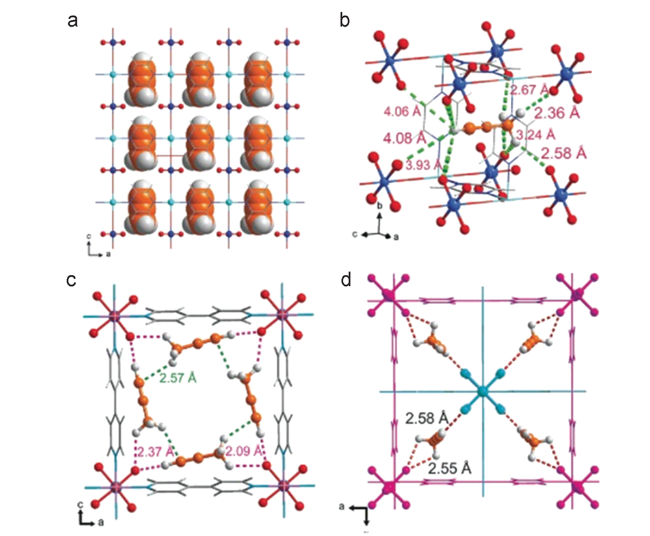
图7 (a,b) DFT-D计算了SIFSIX-3-Ni中C3H4吸附结合位点。(c) DFT-D计算了SIFSIX-1-Cu的C3H4吸附结合位点。(d) DFT-D对SIFSIX-2-Cu-i的C3H4吸附位点进行优化计算(不同的网以粉色和蓝绿色表示)。(颜色代码:F,红色;Si,浅蓝色;C,灰度-40%;H,灰度-25%;N,天蓝色;铜,紫色;Ni,亮绿色。C3H4分子中C原子的颜色用橙色突出显示)[58]Fig. 7 (a,b) DFT-D calculated C3H4 adsorption binding sites in the SIFSIX-3-Ni. (c) DFT-D calculated C3H4 adsorption binding sites in the SIFSIX-1-Cu. (d) DFT-D calculated optimized C3H4 adsorption sites of the SIFSIX-2-Cu-i (The different nets are highlighted in pink and turquoise). (Color code: F, red; Si, light blue; C, gray-40%; H, gray-25%, N, sky blue; Cu, lavender; Ni, bright green. The color of the C atoms in the C3H4 molecule is highlighted by orange) [58] |

图8 (a, b) ELM-12中两种空腔的示意图(Ⅰ和Ⅱ) (Cu,绿色;C,灰色;O,红色;S,黄色;F,亮绿色)。(c, d) 中子衍射晶体结构显示了C3D4分子(位点Ⅰ和Ⅱ)的优先结合位点及其与骨架的紧密接触[61]Fig. 8 (a, b) Schematic diagrams of the two types of cavities (Ⅰ and Ⅱ) in ELM-12 (Cu, green; C, gray; O, red; S, yellow; F, light green). (c, d) Neutron diffraction crystal structure of ELM-12⊃C3D4 showing the preferential binding sites for C3D4 molecules (sites Ⅰ and Ⅱ) and their close contacts with the framework[61] |
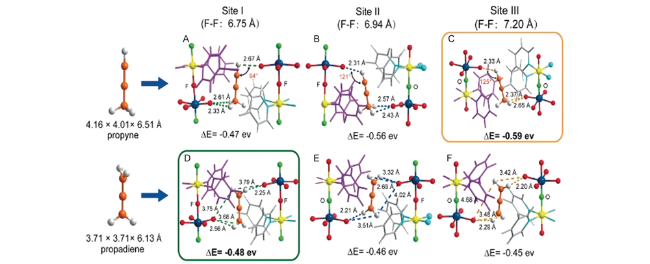
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
图9 ZU-62中丙炔和丙二烯的DFT-D计算结果。F,红色;Nb,深蓝色;C,灰度-40%;H,灰度-25%;O,亮绿色;N,青绿色;铜,黄色;F-F距离,范德华半径[63]Fig. 9 The DFT-D calculated results of the propyne and propadiene within ZU-62. F: red, Nb: dark blue; C: gray-40%, H: gray-25%, O: bright green, N: turquiose, Cu: yellow; the F-F distance includes the Van der Waals radius[63] |