



Contents
1 Introduction
2 Total synthesis of C-20 non-oxygenated ent-kauranes
2.1 Asymmetric total synthesis of(+)-1α,6α-diacetoxy-ent-kaura-9(11),16-dien-12,15-dione and(+)-lungshengenin D by Ma group
2.2 Asymmetric total synthesis of pharicins A~C, and(+)-7-O-acetylpharicin C by Ding group
3 Total synthesis of C-20 oxygenated ent-kauranes
3.1 Total synthesis of maoecrystal P by Luo group
3.2 Total synthesis of(±)-eriocalyxin B,(±)-neolaxiflorin L and(±)-xerophilusin I by Lee group
4 Total synthesis of seco-ent-kauranes
4.1 Total synthesis of(±)-sculpomeatin N by Zhai group
4.2 Total synthesis of(±)-sculpomeatin N by Thomson group
4.3 Total synthesis of(±)-trichorabdal A and(±)- maoecrystal Z by Liang group
4.4 Asymmetric total synthesis of(-)-enmein,(-)-isodocarpin and(-)-sculponin R by Dong group
4.5 Asymmetric total synthesis of(+)-londirabdiol,(-)-longirabdolactone and(-)-effusin by Li group
5 Total synthesis of nor or rearranged-ent-kauranes
5.1 Total synthesis of(±)-jungermannenones B and(±)-jungermannenones C by Lei group
5.2 Asymmetric total synthesis of(-)-maoecrystal V by Baran group
5.3 Asymmetric total synthesis of(+)-jungermatrobrunin A by Lei group
5.4 Asymmetric total synthesis of(+)-ent-kauradienone and(+)-jungermannenones C by Lei group
6 Conclusion and outlook
1 引言
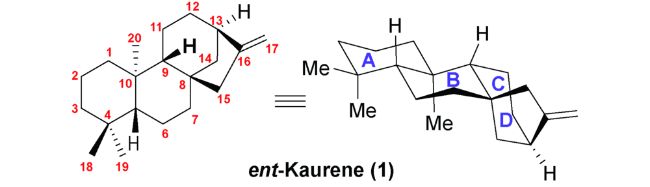
对映-贝壳杉烷二萜类化合物是首次从新西兰产的南洋杉科植物南方贝壳杉的叶精油中发现的一类天然产物[1]。由于该植物在新西兰被称为Kauri pine,故后来人们将这一类具有负的比旋光度的二萜烯称为对映-贝壳杉烯(ent-kaurene)(图式1)。这类天然产物被广泛认为是陆生植物四环二萜中种类最为繁多和结构活性最为多样的一个家族。迄今为止,分离鉴定的天然对映-贝壳杉烷二萜化合物已超过了1500种。他们绝大多数来自于唇形科香茶菜属(Isodon)以及菊科(Compositae)、凤尾蕨科(Pteridaceae)、大戟科(Ephorbiaceae)和苔藓(Livewort)等植物,也有少量来自于微生物代谢产物。我国中科院昆明植物所的孙汉董和普诺·白玛丹增在香茶属植物中对映-贝壳杉烷二萜的分离鉴定和生物活性测试方面做了大量基础性工作[2]。近年来研究表明,该类天然产物中不少化合物具有比较强的抗菌和抗肿瘤活性[3]。
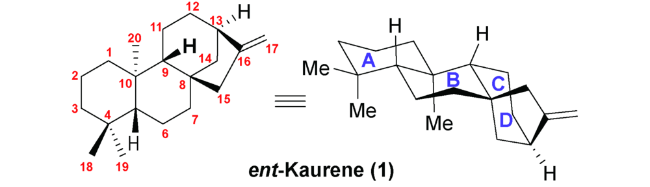
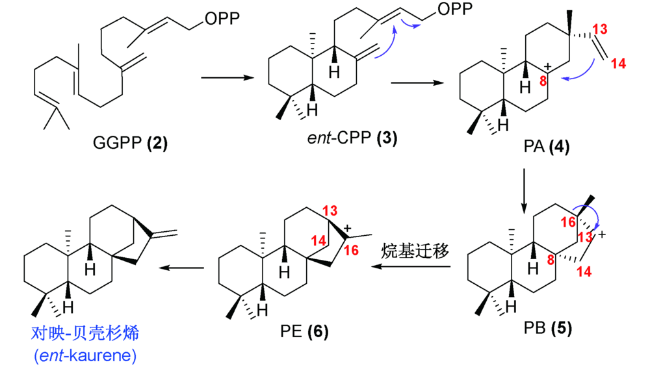
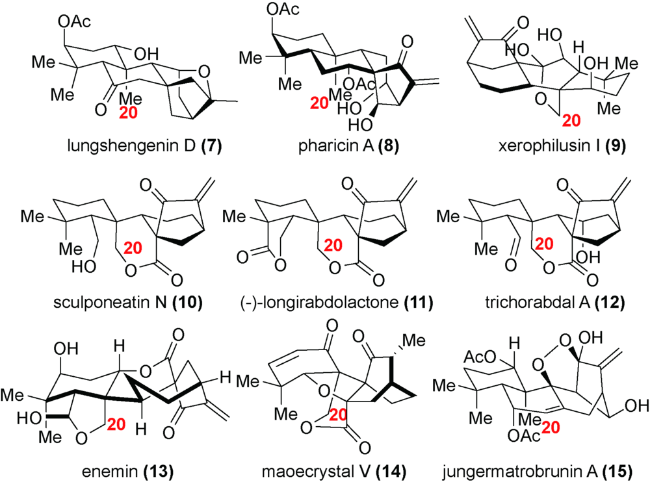
根据结构类似性和分子骨架的不同,对映-贝壳杉烷二萜主要可以分为 C20 位未被氧化的贝壳杉烷(如 lungshengenin D(7))、 C-20 位被氧化的贝壳杉烷(如xerophilusin I(9))、断裂的对映-贝壳杉烷二萜(如 sculpomeatin N(10)),降解或重排的对映-贝壳杉烷二萜(如 maoecrystal V(14))等多种类别(图式3)。由于这类分子在生物活性和结构上的多样性, 对这类分子的全合成研究引起了国内外合成化学家的广泛关注。
本文将会对贝壳杉烷型, 特别是对映-贝壳杉烷型二萜分子的相关合成工作进行总结。鉴于之前雷晓光小组发表的关于对映-贝壳杉烷型天然产物的综述[5]之后,再没有关于此类分子全合成的综述,因此,我们希望在这里介绍2014年以后此类分子的合成工作。
2 C20 位未被氧化的贝壳杉烷
在所有对映-贝壳杉烷二萜类型中,该类二萜是数目最多分布最广的一类,约占整个家族的1/3。其结构特点是C20位始终是一个孤立的甲基,C5和C9位氧化取代的也较少。从香茶菜属植物中得到的该类化合物大多具有高度氧化态,且C15常被氧化,而从非香茶菜属植物中分离的此类化合物氧化度则较低。
2.1 马大为小组对(+)-lungshengenin D 和(+)-1α,6α-diacetoxy-ent-kaura-9(11),16-dien-12,15-dione的不对称全合成
他们从乙氧基环己烯酮出发,引入侧链,随后进行钯催化的不对称脱羧烯丙基化构建全碳的手性季碳中心,得到87% ee的手性烯酮化合物17。随后利用醋酸钯催化的氧化Heck反应成功得到手性的[3.2.1]桥环化合物20,再经过数步转化得到手性醛片段21。另一手性片段的合成是从α甲基环己烯酮化合物24出发,经过CBS不对称还原得到99% ee的手性烯丙醇23,随后与二异丙基氨基甲酰氯缩合得到化合物22。两个手性片段22和21通过Hoppe教授的发展的Homoaldol反应成功实现对接,随后在三氟化硼乙醚介导下发生全新的Mukaiyama-Michael类环化反应,成功实现四环化合物26的制备(图式4)。
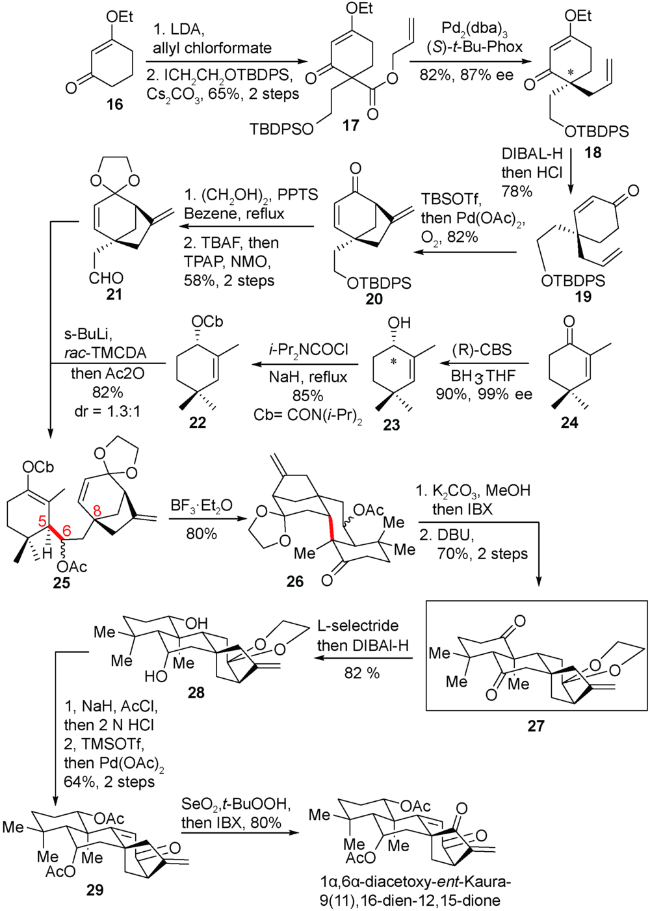
从化合物26出发,通过脱保护、氧化、对C5位的氢进行异构成功得到具有C20位未氧化的对映-贝壳杉烷二萜的母核骨架的高级中间体27。从高级中间体27出发对双羰基进行接力还原、乙酰化,随后对[3.2.1]桥环片段进行氧化态调整便完成一个对映-贝壳杉烷二烯酮的合成(图式4)。
同时从高级中间体27出发,他们对双羰基进行烯醇硅醚化,然后对于位阻较小的部分进行选择性的DDQ氧化,脱除另外一个硅醚就得到烯酮化合物30。随后在C12位羰基α位引入羟基,酸性条件下与环外双键加成关上五元醚环得到化合物32。最后通过数步氧化态的调整,完成了lungshengenin D的不对称合成(图式5)。
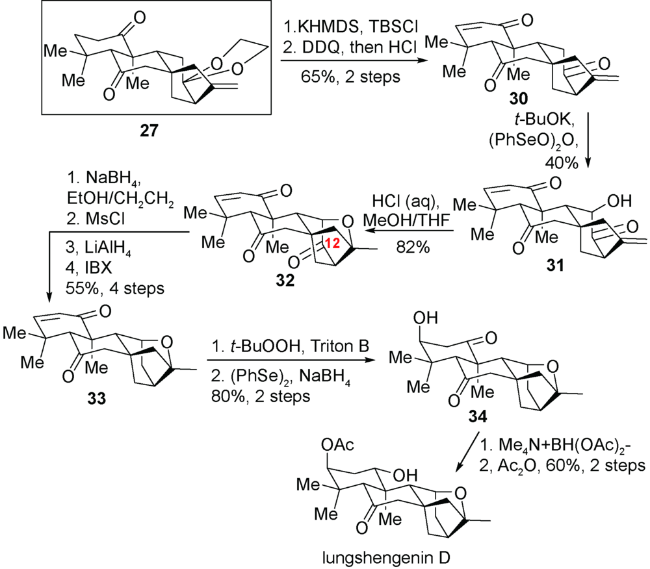
2.2 丁寒锋小组对pharicins A~C 和7-O-acetyl- pharicin C的不对称全合成
如图式6所示,已知化合物Wieland-Miescher 酮35经过6步简单转化即可得到化合物36。化合物36经过Eschenmoser-Tanabe裂解反应得到C5位、C10位分别有侧链的化合物37。化合物37通过几歩简单转化之后得到的环加成前体39在二(三氟乙酸)碘苯的作用下发生氧化去芳构化[5+2]环加成反应实现C9-C8、C11-C12的连接得到中间体41,而后发生频哪醇类型的1,2-酰基迁移反应实现C14-C12的断裂以及C14-C13的连接得到对映-贝壳杉烷骨架42。最后通过对不同部位的氧化态的调整,即可实现天然产物pharicins A~C 和7-O-acetyl-pharicin C的合成。
3 C20 位被氧化的贝壳杉烷
该类二萜与C20未氧化的对映-贝壳杉烷二萜相比,其20位通常为一个氧化的亚甲基、醛基、羧基,并与C3、C4、C7、C11或C14构成(半)缩醛(酮),内酯或醚键氧桥的结构。
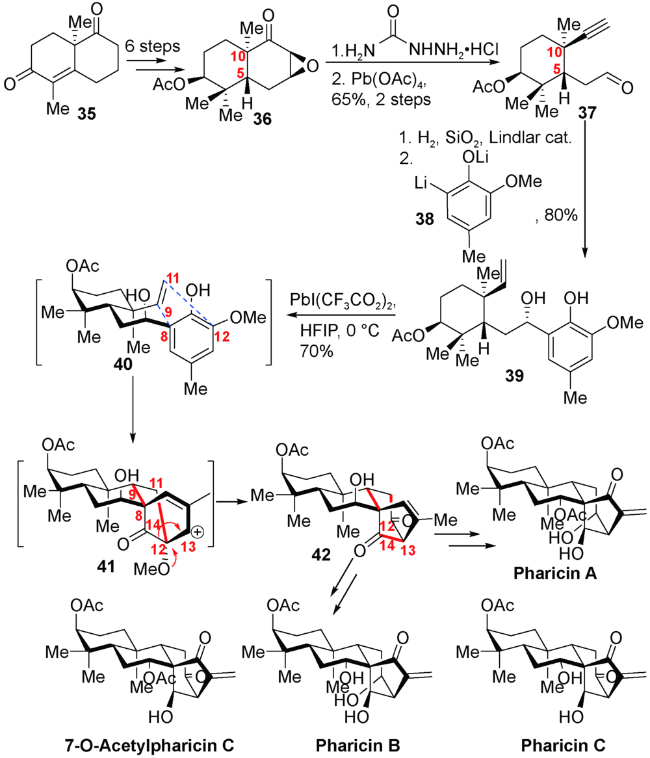
3.1 罗佗平小组对maoecrystal P的全合成
他们从α-甲酰基环己烯酮化合物43出发,通过与双烯体44发生分子间Diels-Alder反应迅速构建A/B顺式并环的四环体系,随后通过羰基还原、生成的羟基进行乙酰基保护、烯丙位溴化后水解氧化得到烯酮化合物47,紧接着数步转换得到关环前体50。在二碘化钐的作用下,化合物50的醛基与烯酮成功实现关环得到具有 [3.2.1]桥环的四环高级中间体51。随后氧化C15位的羟基并引入C16位的亚甲基得到化合物53。m-CPBA氧化烯醇硅醚,酸解脱除保护得到三醇化合物54。紧接着TBAF对C5位氢进行异构,对A环氧化态调整,最终完成C20位氧化的对映-贝壳杉烷二萜maoecrystal P的消旋合成(图式7)。
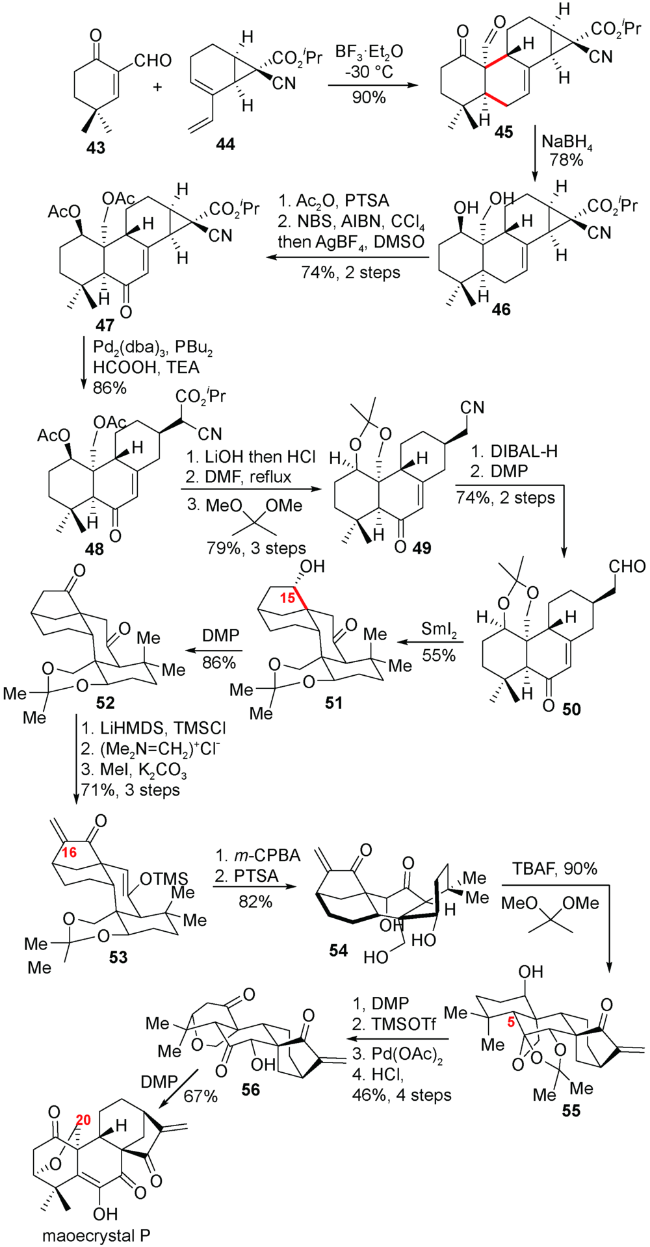
3.2 李志成小组对(±)-eriocalyxin B,(±)-neolaxiflorin L和(±)-xerophilusin I的全合成
该小组从多烯化合物57出发,通过与α甲酰基环己烯酮化合物43发生Diels-Alder反应得到A/B顺式并环的化合物58。随后羰基还原、生成的羟基进行硅基保护,氧化得到关环前体60。在筛选大量Lewis酸后,作者发现在溴化锂和二甲基氯化铝促进下,能够实现Mukaiyama-Michael/carbocyclization串联环化反应,仅以5步便能高效地得到具有C20位氧化的对映-贝壳杉烷二萜的母核骨架61(图式8)。
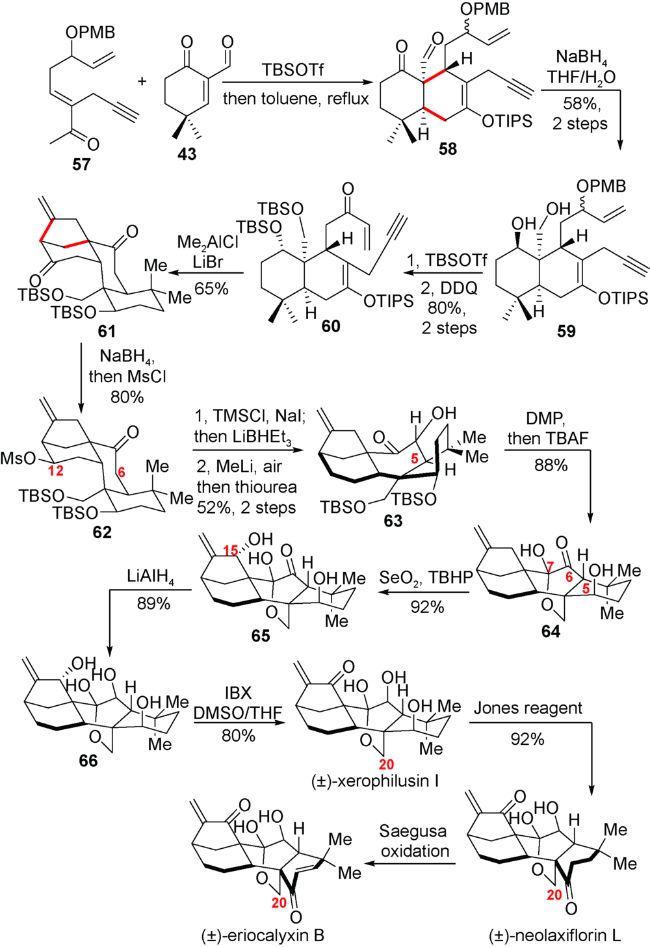
随后他们对中间体61的C12位羰基进行消除,再引入C6位的氧化态就得到化合物63。紧接着DMP氧化,TBAF脱除硅基保护,成功关上C20与C7的半缩酮环并且实现C5位氢的异构。生成的中间体64进行Riley氧化引入C15位氧化态,最后对生成的化合物65进行LiAlH4还原C6位羰基,以及A环氧化态调整完成了eriocalyxin B, neolaxiflorin L 和xerophilusin I的消旋合成。
4 断裂的对映-贝壳杉烷二萜
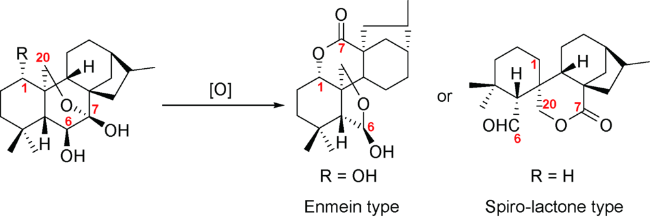
从生源合成上看,对映-贝壳杉烷二萜化合物中的碳碳键在植物体内发生了氧化断裂就可以生成该类二萜。该类二萜数量较大,主要以C6,C7-断裂为主。按照结构的不同可以分为两大类型:延命素型(Enmein type)和螺环内酯型(Spiro-lactone type)(图式9)。
4.1 翟宏斌小组对(±)-sculpomeatin N的全合成
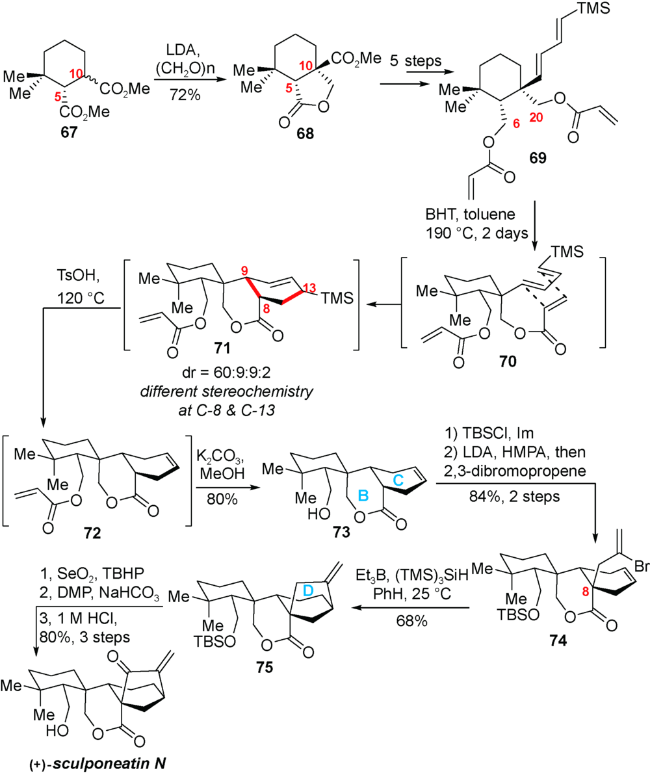
该合成路线以已知化合物67作为起始原料,在LDA的作用下与多聚甲醛发生aldol反应,生成的醇负离子原位进攻分子内的酯基发生酯交换反应生成内酯化合物68,该步反应成功确立了C5、C10位的相对构型。化合物68通过几歩转化即可得到Diels-Alder反应前体69。在BHT的存在下,以甲苯作为溶剂190 ℃下反应两天得到以endo产物71为主的Diels-Alder产物(dr=60∶9∶9∶2)。该中间体不经分离,直接向反应体系中加入PTSA脱除TMS,而后加入K2CO3与甲醇脱除酯基就得到醇73。接着将化合物73裸露的羟基进行保护之后引入C8侧链,通过自由基环化反应构建D环得到化合物75。最后通过Riley氧化、DMP氧化以及脱硅基保护,成功实现天然产物sculpomeatin N的合成(图式10)。
4.2 Thomson小组对(±)-sculpomeatin N的全合成
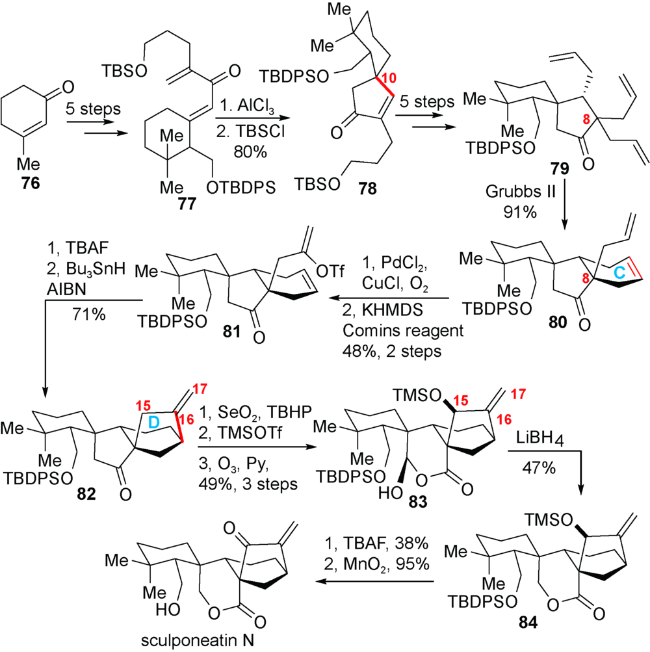
如图式11,他们以廉价易得的3-甲基-2-环己烯-1-酮76为原料,通过5步转化得到Nazarov环化反应前体77。化合物77在AlCl3作用下发生Nazarov环化反应成功构建了所需的C10构型,而后对裸露的羟基进行保护得到化合物78,两步反应的总收率为 80%。在对化合物78进行Diels-Alder反应直接构建C环的尝试失败之后,他们计划采取迂回的思路,利用分子内烯烃复分解反应来构建C环。于是,他们将化合物78通过5步转化引入侧链得到化合物79,在Grubbs II催化下发生分子内烯烃复分解反应,以91%的收率得到期望的产物化合物80。化合物80的末端双键发生Wacker氧化后,生成的甲基酮在KHMDS的作用下与Comins试剂反应得到三氟甲磺酸烯醇酯81。在对化合物81的分子内还原Heck反应尝试受阻之后,他们发现化合物81在TBAF作用下发生消除反应得到的炔烃可发生自由基介导的还原环化反应构建关键的[3.2.1]桥环,得到化合物82,两步反应的总收率为 71%。至此,化合物82与目标天然产物sculpomeatin N相比,只剩下最后的内酯环。他们计划通过臭氧化断裂烯醇硅醚而后还原生成的醛基实现内酯环的构建。为了预防C16-C17环外双键在臭氧下发生竞争反应,因此先通过Riley氧化引入C15羟基,以期利用诱导效应降低C16-C17环外双键的反应活性。接下来,利用TMSOTf生成烯醇硅醚的同时,对 C15羟基进行硅基保护,而后顺利进行臭氧化反应,以3步49%的收率得到内半缩醛83。化合物83与LiBH4反应,以47%的收率将内半缩醛还原成内酯84。最后,通过TBAF脱去硅基保护,利用二氧化锰氧化生成的烯丙醇,即可以两步36%的收率得到天然产物sculpomeatin N。
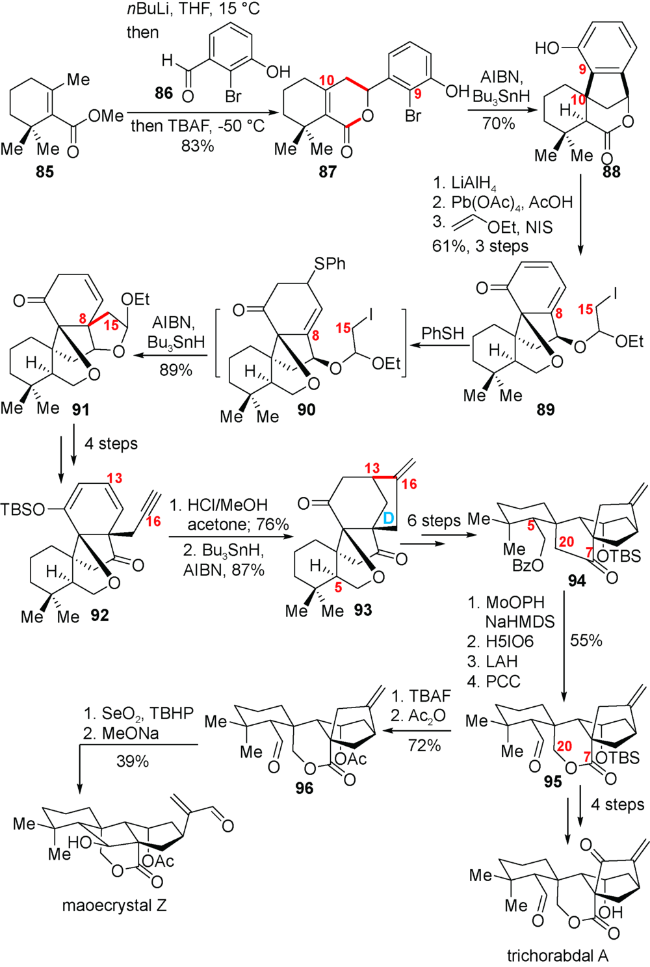
4.3 梁广鑫小组对(±)-trichorabdal A和(±)- maoecrystal Z的全合成
如图式12所示,他们从已知的化合物85出发,通过酯基γ位甲基与醛86发生aldol反应生成内酯87。87在AIBN与 Bu3SnH的存在下发生自由基环化反应连接C9-C10位化学键,以70%的收率得到化合物88。化合物88在LiAlH4作用下生成的二醇在去芳构化反应条件下得到的化合物可以顺利地转化为Ueno-Stork环化反应的前体89。梁广鑫小组对Ueno-Stork环化反应进行了大量的尝试,但实验均以失败告终。考虑到较为敏感的1,6-烯酮结构是反应失败的主要原因,他们就先利用苯硫酚进攻烯酮发生1,4-加成来破坏1,6-烯酮结构,而后进行Ueno-Stork环化反应尝试。最后他们发现可以连接C8-C15而构建C8位季碳中心,以89%的收率得到环化产物91。化合物91经过几歩简单的转化可以得到环化前体92。化合物92在盐酸作用下生成的α, β-不饱和酮可以被末端炔烃在AIBN与 Bu3SnH的作用下生成的自由基进攻,成功连接C13-C16构建D环得到化合物93。化合物93通过氧化还原以及保护基操作,实现了C5位构型的异构化,得到化合物94。化合物94羰基的α位引入羟基之后,在高碘酸的作用下发生氧化断裂得到内半缩醛,而后利用LiAlH4还原实现内半缩醛到内酯转化的同时脱去Bz保护,生成的伯醇被PCC氧化得到高级中间体95。化合物95通过简单几步转化即可实现天然产物trichorabdal A的合成。另一方面,对于天然产物maoecrystal Z,作者希望通过逆aldol/aldol反应实现。即化合物95先将TBS转化为乙酰基,而后Riley氧化生成的烯丙醇在碱性条件下发生所期盼的逆aldol/aldol反应,成功实现了天然产物maoecrystal Z的合成。这个结果也佐证了trichorabdal A与maoecrystal Z潜在的生源合成联系。
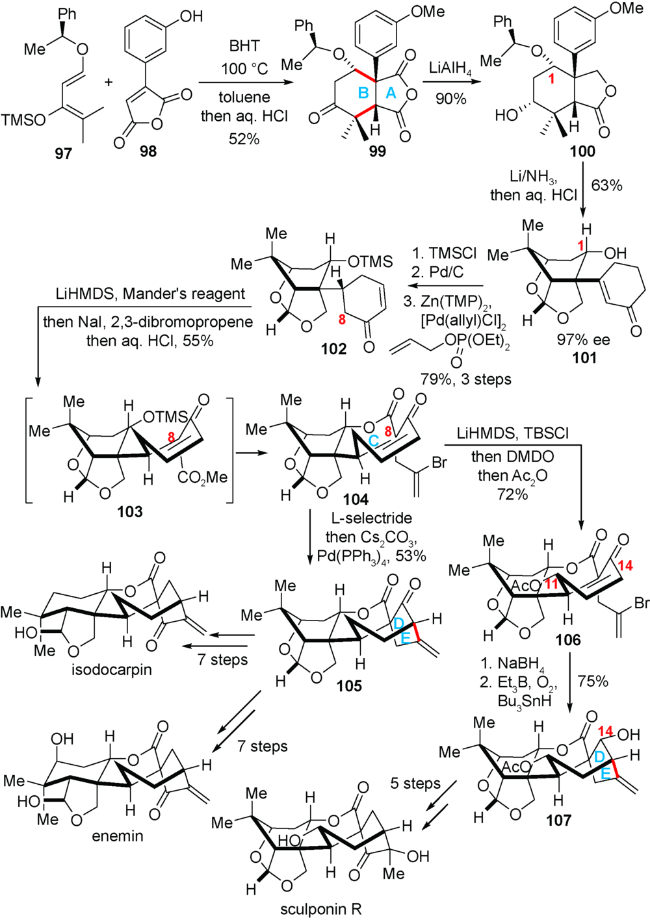
4.4 董广斌小组对(-)-enmein,(-)-isodocarpin和(-)-sculponin R的全合成
2018年,美国芝加哥大学的董广斌小组报道了天然产物(-)-enmein,(-)-isodocarpin与(-)-sculponin R的全合成研究[24]。该合成路线前期通过Diels-Alder反应快速构建AB环,接下来利用一锅法酰化/烷基化/内酯化反应构建C环以及C8季碳中心,最后利用还原烯基化反应实现D/E环的构建,以实现三个延命素型天然产物的发散式合成。
如图式13所示,已知的手性双烯体97与酸酐98发生Diels-Alder反应,在盐酸淬灭后,可以52%的收率得到endo构型为主的加成产物99。化合物99在LiAlH4的作用下,在选择性地还原酸酐中一个羰基的同时,也还原了酮羰基得到化合物100。化合物100在Li/NH3的作用下进行了三次转化:1)苯环发生Birch还原得到1,4-环己二烯;2)内酯发生还原得到半缩醛;3)移除了C1位的苄基保护基;而后在酸性条件下淬灭,1,4-环己二烯转化为环己烯酮,同时半缩醛转化为笼状缩醛,得到化合物101。化合物101进行3步转化得到的α, β-不饱和酮102。在羰基α位引入甲酯之后,102再次与2,3-二溴丙烯反应引入第二条侧链而构建C8位季碳中心。生成的中间体在酸性条件下淬灭即可构建内酯环C环,得到关键高级中间体104。化合物104制成烯醇硅醚后通过原位环氧化开环和乙酰基保护,引入C11位氧化态得到化合物106。化合物106在还原C14羰基之后,发生自由基环化反应,构建[3.2.1]桥环,得到化合物107。化合物107通过几歩简单转化即可实现天然产物(-)-sculponin R的全合成。而另一方面,化合物104和L-selectride发生1,4-还原,生成的烯醇锂盐在Cs2CO3与Pd(PPh3)4的作用下发生钯催化的烯基化反应构建[3.2.1]桥环,得到化合物105,进而通过几步官能团操作实现天然产物(-)-enmein和(-)-isodocarpin的全合成。
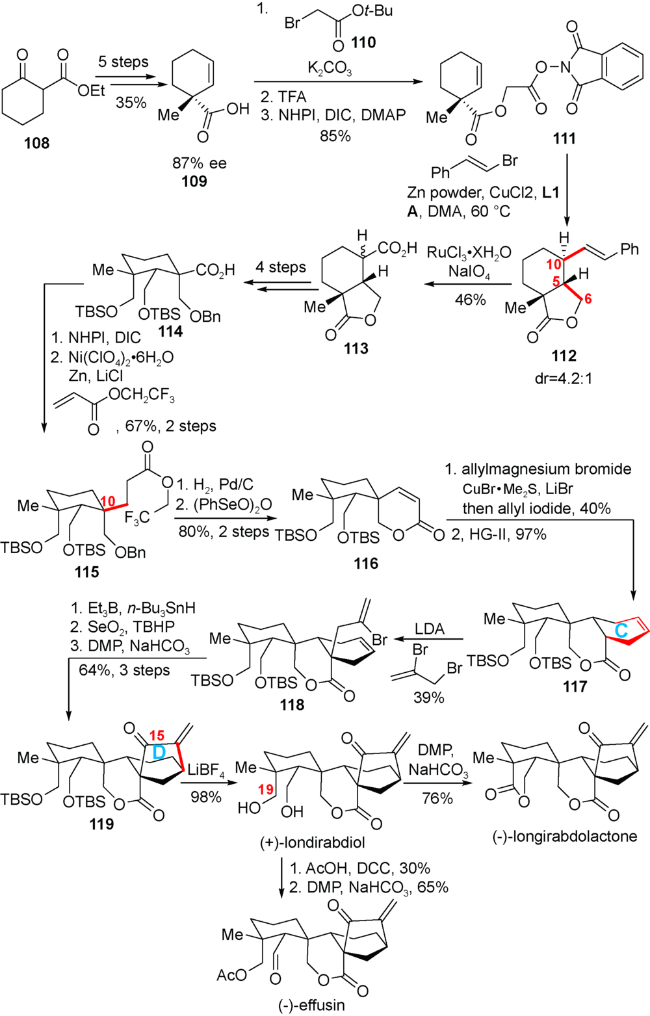
4.5 李超小组对(+)-londirabdiol,(-)-longirabdolactone和(-)-effusin的全合成
如图式14所示,他们以廉价易得的2-环己酮甲酸乙酯108为原料,通过5步反应转化得到光学纯度为87% ee的化合物109,再对羧基进行衍生化,以3步85%的收率得到脱羧前体111。接下来他们详细考察了中间体111的自由基脱羧环化烯基化反应,并最终发现该反应可在氯化铜的催化下,以DavePhos作为配体L1,2-溴丙酸作为添加剂A,与锌粉和溴代烯在DMA中60 ℃下反应得到化合物112。该反应在连接C5-C6碳碳键以及引入C10侧链的同时,成功地利用C4位手性控制所需的C5位构型。化合物112的双键发生氧化断裂得到羧酸113,两步反应的总收率为 46%。化合物113通过还原内酯环而后进行保护基操作可得到羧酸114。羧酸114转化为氧化还原活性酯之后,与2,2,2-三氟乙基丙烯酸酯发生脱羧Giese反应得到化合物115,在引入C10位的第二条侧链的同时,利用底物构型很好地控制了C10位的手性中心,两步反应的总收率为67%。化合物115脱除苄基保护之后自发生成内酯,然后在苯亚硒酸酐的作用下可得到不饱和内酯116。接下来他们构建C环的策略与Thomson小组类似。即利用烯丙基铜锂试剂发生1,4-加成,原位与烯丙基碘反应引入两条侧链,而后发生分子内的烯烃复分解反应,以两步39%的收率得到化合物117。化合物117在酯基α位引入侧链后发生自由基环化反应构建D环,而后引入C15位氧化态得到化合物119。化合物119利用LiBF4脱除硅基保护之后即可得到天然产物(+)-londirabdiol。(+)-londirabdiol在DMP作用下氧化二醇可得到天然产物(-)-longirabdolactone。而如果将(+)-londirabdiol的C19位羟基进行乙酰化之后进行DMP氧化,即可实现天然产物(-)-effusin的全合成。
5 降解或重排的对映-贝壳杉烷二萜
该类化合物的骨架结构独特,很多甚至无法简单辨认出其原有的对映-贝壳杉烷二萜骨架。一般而言,该类分子是在贝壳杉烷骨架的基础上由新的碳碳键生成或碳碳键发生迁移重排形成的,伴随着碳原子的降解,可以得到结构各异的天然产物。
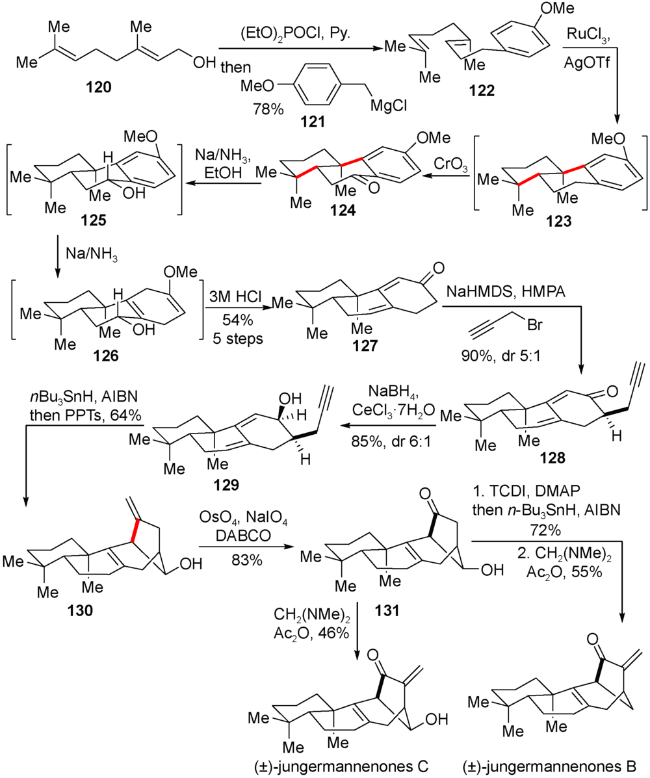
5.1 雷晓光小组对(±)-jungermannenones B 和(±)-jungermannenones C的合成
2015年,雷晓光小组报道了重排型的对映-贝壳杉烷jungermannenones B 和 C的消旋合成[28]。在合成中,他们的策略是采用阳离子串联环化[29]构建三环体系,Birch还原引入炔丙基侧链,以及自由基环化关上重排的[3.2.1]桥环。该小组从廉价易得的香叶醇120出发,经格氏试剂进攻,得到芳香双烯化合物122,随后再经三价钌催化串联环化紧接着三氧化铬苄位氧化得到三环化合物124。Birch还原取代苯环,酸解以后在得到的二烯酮产物127上引入炔丙基侧链生成128,在128的羰基被还原成醇129以后,进行三丁基锡氢自由基环化构筑关键的[3.2.1]桥环片段而得到130。最后通过几步氧化态调整最终完成jungermannenones B 和 C的消旋合成(图式15)。
5.2 Baran小组对(-)-maoecrystal V的合成
该小组从环己烯酮132出发,通过手性铜试剂的不对称Micheal加成顺利引入烯基片段而得到134,在134的羰基α位引入氧化态后,生成的化合物135在乙基二氯化铝的活化下经历反Baldwin环化成功实现[3.2.1]桥环的构建得到化合物136。紧接着羰基化合物137与烯基碘138发生aldol反应,成功地实现两个片段对接而得到139。随后139在酸性条件下发生频哪醇重排以及双键异构实现[3.2.1]桥环向[2.2.2]桥环的转化,得到化合物140。在筛选大量Lewis酸之后,发现只有在氯化镧和氯化锂的作用下能够成功的在C10位置引入羟甲基而实现C10位全碳季碳的构筑,得到化合物141。随后通过大量还原剂的尝试,发现在三氟甲磺酸锌与硼氢化锂原位产生的的硼氢化锌作用下,可以成功完成C5位羰基立体选择性的还原得到羟基构型正确的化合物143。紧接着通过几步保护基调整,氰基引入与水解得到内酯化合物146,随后通过环氧化-开环,氧化消除,最终以11步完成(-)-maoecrystal V的不对称合成(图式16)。
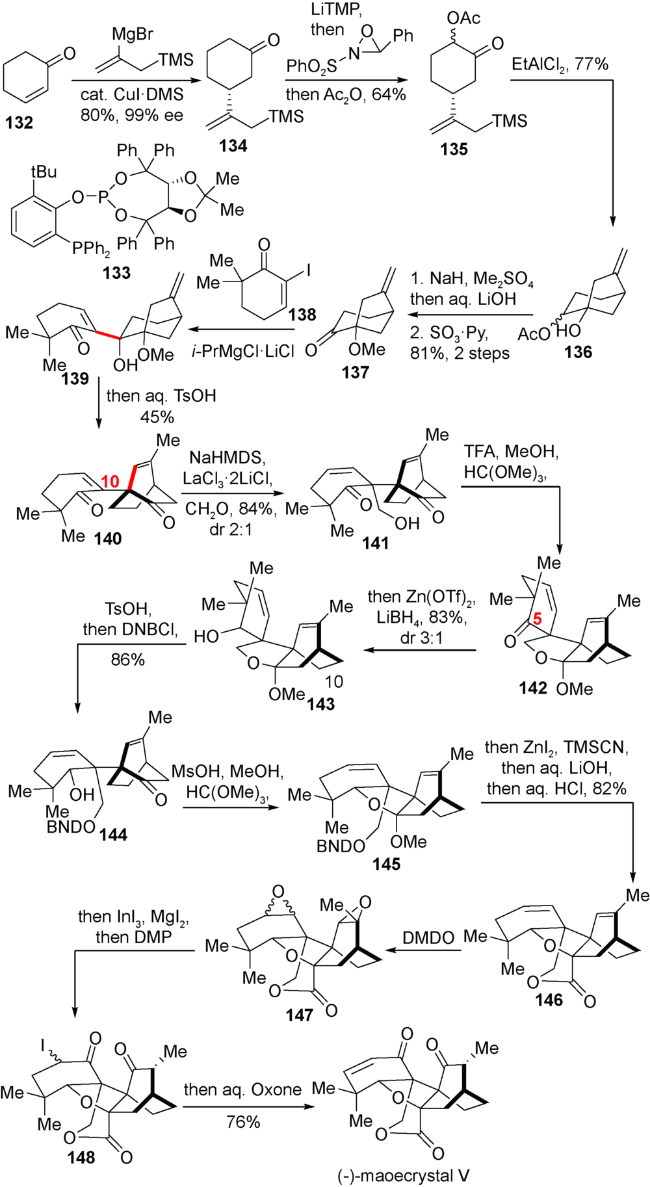
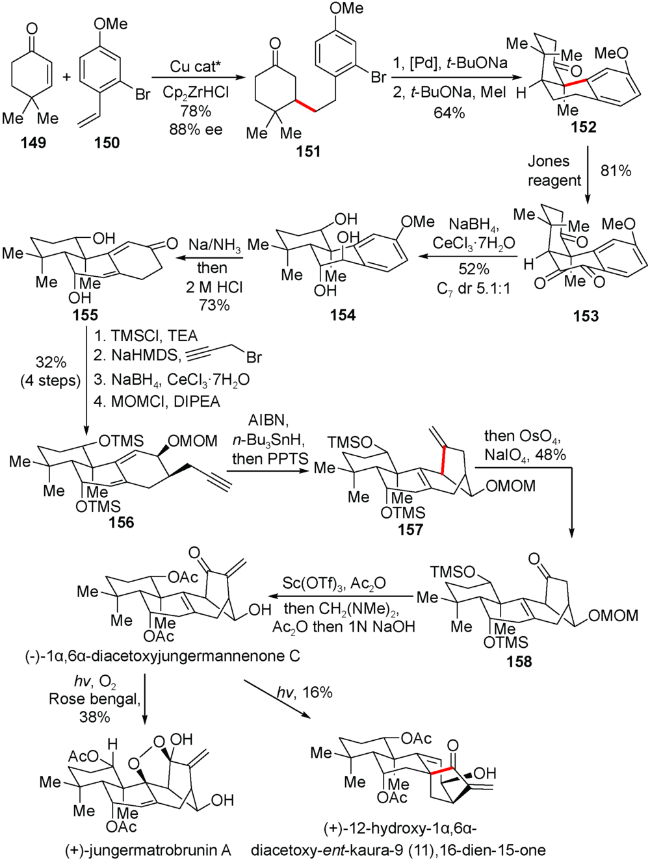
5.3 雷晓光小组对(+)-jungermatrobrunin A的合成
该小组利用芳基乙烯150在锆氢参与手性铜试剂催化下与环己烯酮化合物发生1,4不对称加成得到光学纯度为88% ee的化合物151。随后利用钯催化的芳基溴代物与酮基碳负离子进行的偶联反应,以及接下来的甲基化反应得到关环化合物152,三氧化铬氧化152的苄位以及高苄位得到双酮化合物153。在Luche还原之后进行二醇154的Birch还原得到双烯酮化合物155。然后引入炔丙基侧链,通过156的自由基环化得到关键重排的[3.2.1]桥环结构157。随后通过氧化态调整引入亚甲基完成天然产物(-)-1α,6α-diacetoxyjungermannenone C的合成。最后在光照下与氧气发生Schenck ene反应实现天然产物(+)-jungermatrobrunin A的不对成合成。同时还发现光照下重排的[3.2.1]桥环结构能够与对映-贝壳杉烷的[3.2.1]桥环结构相互转化,并完成(+)-12-hydroxy-1α,6α-diacetoxy-ent-kaura-9(11),16- dien-15-one的合成(图式17)。
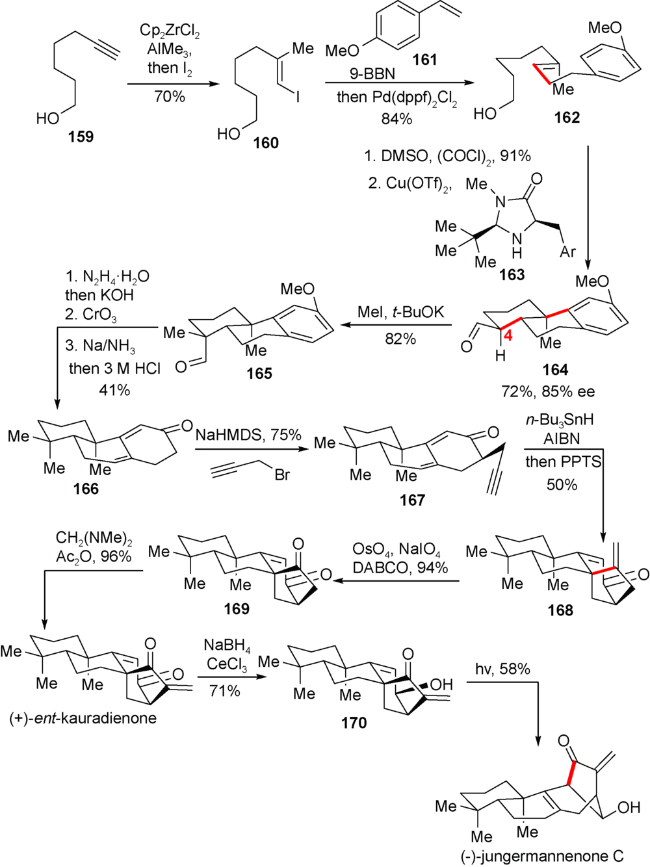
5.4 雷晓光小组对(+)-ent-kauradienone和(+)-jungermannenones C的合成
2019年,雷晓光小组在完成重排型的对映-贝壳杉烷(+)-jungermatrobrunin A的基础之上实现对映-贝壳杉烷分子的骨架向重排型的对映-贝壳杉烷骨架的转化并完成jungermannenones C的不对称合成[34]。在合成中,他们在之前的研究基础之上发现光照下重排的[3.2.1]桥环结构能够与对映-贝壳杉烷的[3.2.1]桥环结构相互转化,利用这一发现他们优化了jungermannenones C的合成。
该小组从炔醇159出发经过2步转换得到芳基链状醇162,Swern氧化162的羟基之后,利用MacMillan催化剂163经历SOMO活化发生分子内串联环化二得到具有85% ee的手性化合物164。随后引入C4位的甲基,醛基脱氧,苄位氧化以及Birch还原得到双烯酮化合物166。在引入炔丙基侧链以后,炔基在三丁基锡氢作用下产生烯基自由基并对烯酮进攻得到具有对映-贝壳杉烷骨架的四环高级中间体168。随后氧化断裂,亚甲基引入便得到天然产物(+)-ent-kauradienone。最后Luche还原,在光照下实现对映-贝壳杉烷的[3.2.1]桥环结构向重排的[3.2.1]桥环结构的转化,最终完成(-)-jungermannenones C的不对称合成(图式18)。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
6 结论与展望
作为四环二萜体系的代表,对映-贝壳杉烷二萜因其良好的生物活性和新颖的骨架结构,在过去半个世纪一直受到合成化学家的关注。早期合成路线的线性步骤一般都在25步以上并且仅有数个氧化态比较低的二萜被合成。随着近年来骨架更为奇特、生物活性更为多样的对映-贝壳杉烷类二萜分子被分离鉴定,越来越多合成化学家被这个领域所吸引,并为实现这类分子简洁高效的合成发展了许多新颖的策略。迄今为止,已有20余种具有不同骨架类型的对映-贝壳杉烷二萜的全合成实现,个别明星分子的合成路线还被反复刷新和简化。2015年以前,对映-贝壳杉烷的合成主要由国外的合成课题组研究,而国内的合成化学家对于此类分子的合成并未给予较大关注。2015年以后,这一类分子的合成热潮逐渐向国内转移,此间一批优秀的中国合成化学家开展这一家族各类骨架分子的合成研究。仅2017年至今,国内化学研究者就有9篇关于对映-贝壳杉烷家族分子的合成报道发表。这些研究不仅能够催生出一些高效、简洁和可放大的合成策略,同时也可以对一大批相关的天然产物及其类似物进行深入的生物活性研究。而这些生物活性研究也可以为这个家族的天然产物的结构-活性关系提供有用的信息,促进这些化合物的作用机制的研究以及基于这些天然产物的工具药物和疾病治疗药物的发现。