




Contents
1 Introduction
2 Synthesis of cyclic carbonate from CO2 and epoxide catalyzed by MOFs
2.1 Monometallic MOFs node for Lewis acid catalysis
2.2 Metal cluster MOFs node for Lewis acid catalysis
2.3 Metalloligand in MOFs for Lewis acid catalysis
2.4 Double catalytic sites in MOFs
3 Carboxylation of CO2 with terminal alkynes catalyzed by MOFs
4 Cyclization of CO2 with propargylic amide/alcohol catalyzed by MOFs
5 Conclusion
1 引言
近几十年来,随着人类社会和工业的发展,煤炭、石油、天然气等碳氢燃料燃烧产生的CO2,远远超过了过去的水平。由大气中CO2含量增加引起的海洋酸化、全球变暖、生态系统异常等一系列环境问题已受到人们的广泛关注。在21世纪,碳捕获和封存被认为是克服这些问题并对环境保护和可持续发展产生重大影响的战略。另一方面,从资源角度讲,CO2是一种安全丰富的C1资源,开发简易高效的方法吸附和转化大气中的CO2是颇具吸引力的研究领域。目前,已有多种催化剂,包括金属氧化物[1]、分子筛[2]、二氧化硅负载金属盐[3]和多孔聚合物[4],能够以CO2为原料合成有用化工产品。然而,许多因素限制了它们的实际工业应用,包括高成本、低效率、寿命短,可回收性差等。因此,迫切需要开发和研究高效催化剂在温和条件下转化CO2为有用化学品。
金属有机框架(Metal-organic frameworks, MOFs)是利用有机配体和金属离子(或金属簇)之间配位组装形成的具有超分子多孔网络结构的晶态有机-无机杂化材料。基于较高热稳定性、较大比表面积、有序多孔结构、可调孔径大小等特点,近年来MOFs在多相催化[5,6,7,8]、吸附与分离[9]、分子识别[10]、药物传输[11]、质子传导[12]等众多领域展现出潜在的应用前景。在这些功能应用中,多相催化是发展最为迅速的领域之一。MOFs具有节点多样、有机配体可设计、空腔/窗口类型大小可调节等特点,因而框架组装具有无限多样性和可修饰性。不仅如此,MOFs多相催化剂还具有诸多优越性,如空间限域效应[13]、主客体识别效应[14]、立体和电子效应[15]以及框架内两个或者多个催化活性位点的协同效应[16]。此外,MOFs还显示出其他一些重要特性,如较高的CO2吸附性能[17,18,19],可以提高框架内催化活性中心周围CO2浓度,从而提高催化效率。最后,MOFs可以方便地从液相或气相反应中分离出来,实现简单回收和重复利用。目前,MOFs催化剂中活性位点主要分为三类:第一类是以MOFs节点金属离子(金属簇)的开放位点作为活性中心,发挥Lewis酸催化作用;第二类是有机配体中的官能团直接作为活性中心,或者通过后合成修饰引入新的催化中心;第三类是以MOFs为载体,采用吸附、浸渍、还原等方法将活性中心负载到MOFs表面或空腔中。
本文针对近年来以MOFs为多相催化剂在CO2参与的有机合成反应中的应用,如CO2与环氧化物、末端炔、炔丙醇/胺等发生的化学反应,以MOFs催化活性中心为类别作一详细的综述。
2 MOFs催化CO2与环氧化物合成环状碳酸酯
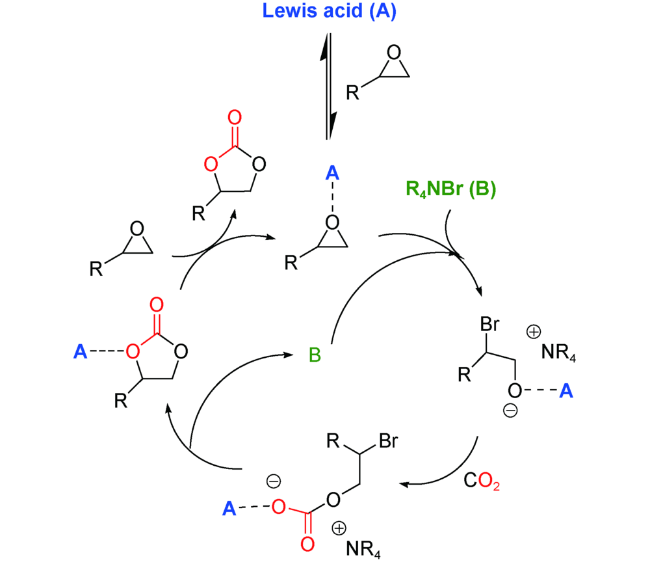
2.1 MOFs节点单金属Lewis酸催化
ZIFs系列是最具代表性的一类以单金属作为节点的MOFs。该材料是以咪唑及其衍生物为桥连配体通过N原子与Zn或Co等金属离子组装形成的具有类似硅铝分子筛孔道结构的沸石咪唑框架。ZIFs通常比其他MOFs材料具有更高的热稳定性和化学稳定性,如将ZIF-8放入水、甲醇中煮沸1~7天仍能保持结构的相对稳定[24],为这类材料的广泛应用奠定了基础。
2011年,Carreon等[25]报道了胺官能化的ZIF-8催化氯代环氧乙烷转化为相应环状碳酸酯的研究。ZIF-8中节点Zn2+呈四配位构型,处于配位不饱和状态,因而可发挥Lewis酸作用,而配体上的咪唑氮原子和框架中引入的乙二胺起Lewis碱作用,两者之间的协同催化促进反应的高效进行,在无共催化剂和溶剂,7 bar CO2和70 ℃条件下,产率可达73%。CO2吸附数据表明,胺官能化的ZIF-8的CO2吸附量高于未修饰过的ZIF-8,因此,他们认为在ZIF-8内引入胺,可以促进CO2在框架内的吸附活化,从而使反应更加高效地进行。遗憾的是,ZIF-8在一次催化循环后失去活性。
2015年,Park等[26]以硝酸锌和2-醛基咪唑为原料合成了ZIF-90,并探索了其在无助催化剂和溶剂条件下催化环氧丙烷和CO2的环加成反应。在120 ℃和CO2压力为1.2 MPa条件下,反应8 h,产率可达88%。他们通过DFT计算,从理论上解释了ZIF-90的催化机理,即ZIF-90中的Zn2+(Lewis酸性中心)与环氧丙烷的氧原子相互作用,活化环氧化物,同时,ZIF-90的醛基活化CO2形成开环中间体,进而关环生成环状碳酸酯。此外,该催化剂能够离心分离,循环使用5次而不会显著损失催化活性。
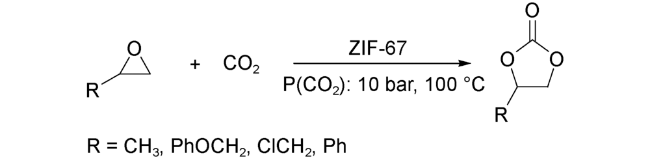
2016年,Park等[27]又采用2-甲基咪唑和硝酸钴在室温下和水介质中快速合成了与ZIF-8同构的ZIF-67。在无溶剂和无共催化剂条件下,ZIF-67可高效催化CO2与环氧丙烷的环加成反应。与含锌的ZIF-8相比,含钴的ZIF-67表现出更好的选择性。此外,ZIF-67稳定性好,可重复使用且无需任何活化步骤。他们认为催化剂外表面的结构缺陷产生了一种特殊的酸-碱性位点,这是ZIF-67具有催化活性的本质,也是所有ZIFs材料具有催化活性的基本原理。
同年,Verpoort等[28]利用ZIF-67实现了一系列环氧化物到环状碳酸酯的高效转化(图式2)。在该反应过程中,他们认为ZIF-67中的Co2+作为Lewis酸性位点活化环氧化物,而咪唑N原子则作为Lewis碱性位点吸附活化CO2。ZIF-67中Lewis酸-碱之间的共同作用,可促使反应在相对温和的条件下高效高选择性地进行,效率远高于之前报道的ZIF-8。
2016年,Nagaraja等[31]利用己二烯二酸分别与1,2-双(4-吡啶基)乙烷、1,2-双(4-吡啶基)乙烯和4,4'-双(偶氮二吡啶)组成混合配体,以乙酸镍为金属盐,合成了一系列具有三重穿插网络的同构型Ni-MOFs。单晶结构分析表明,这类MOFs中节点Ni2+分别与2个羧酸氧原子、2个吡啶氮原子以及2个水分子配位,形成六配位八面体构型。通过高温热处理,与Ni2+配位的两个水分子可以被脱除,使其变为四配位不饱和状态,从而具备Lewis酸催化活性。活化后的Ni-MOF与TBAB共同作用,可在相对温和的条件下(80 ℃和0.8 MPa CO2)催化一系列环氧化物高效转化为环状碳酸酯。循环实验表明,该催化剂在重复使用4次之后,活性未明显降低且结构仍旧保持稳定。
2.2 MOFs节点金属簇Lewis酸催化
(1)Zn4O簇
Zn4O簇是IRMOF(Isoreticular metal-organic frameworks)系列的代表性金属节点,通常为4个Zn2+与1个μ4-O2-构成的Zn-O四面体,其中,每个Zn2+分别与1个μ4-O2-和3个羧酸离子配位,呈现四配位构型,具有潜在的Lewis酸催化活性。1999年,Yaghi等[32]以对苯二甲酸(BDC)和硝酸锌为原料,合成出孔径为1.29 nm的IRMOF-1(MOF-5)。该MOF的框架孔隙率约为60%,比表面积高达2900 cm3/g,实现了晶态微孔材料向晶态介孔材料转变的重要进展。
2009年,Han等[33]以IRMOF-1/TBAB为双组分催化剂,在相对温和条件下(50 ℃和0.1 MPa CO2)高效地合成了环状碳酸酯,且催化剂使用3次后,活性并未明显降低。他们认为较高的催化活性源于MOFs中节点Zn4O金属簇的Lewis酸性以及MOFs框架较强的CO2吸附性能。
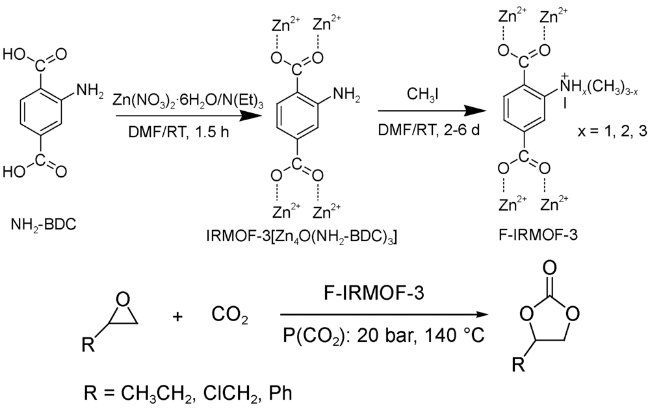
从上述研究可知,IRMOF-1/TBAB的双组分催化体系可以高效地催化环氧化物到环状碳酸酯的转化。然而,体系中的共催化剂季胺盐使得催化剂和产物的分离困难,如能通过催化剂的合理设计避免外加共催化剂的使用,将有效地改善催化体系,实现反应更加绿色化。2012年,Wang等[34]利用碘甲烷对IRMOF-3进行后合成修饰,成功地在MOF框架内固载了季胺盐(图式3)。在功能化后的IRMOF-3中,Zn4O簇中配位不饱和的Zn2+与配体上的季胺盐共同作用,实现了IRMOF-3单组分高效催化环碳酸酯的合成。循环实验证明,该催化剂可以重复使用4次而无明显的活性降低。
2016年,Park等[35]以2-氨基对苯二甲酸和1,3,5-三(4-羧基苯基)苯为混合配体,硝酸锌为金属盐,构建了一例具有微孔-介孔结构的新型MOFs催化剂UMCM-1-NH2,并实现了其在温和条件下(25 ℃和1.2 MPa CO2)高效催化环状碳酸酯类化合物的合成。对比过往的催化体系,他们提出UMCM-1-NH2中的多级孔结构对反应活性起到了关键性的作用,其中,介孔结构有效地促进了底物之间的传质过程,而微孔结构则利于底物和Zn4O簇Lewis酸催化位点充分作用。因此,UMCM-1-NH2可以在室温条件下实现环氧化物到环碳酸酯的完全转化。上述例子表明,对MOFs催化剂的孔结构特性进行修饰,同样可以有效地调控反应活性。
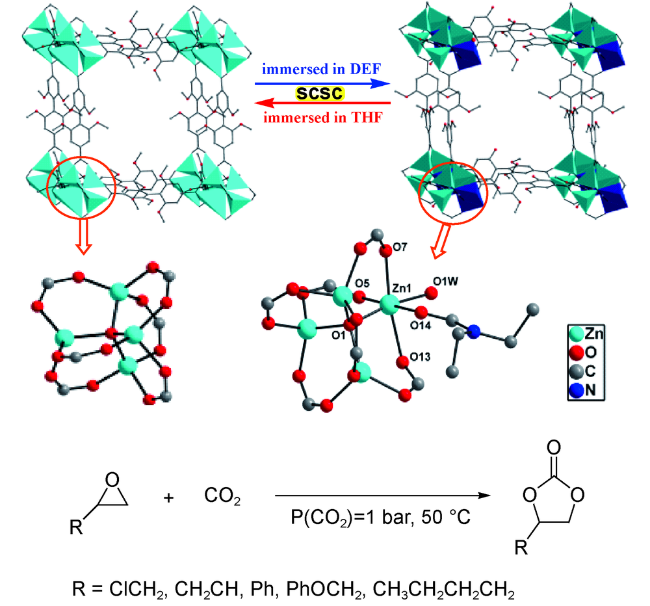
2017年,Ren等[36]利用溶剂诱导法,通过单晶到单晶的方式,实现了两种同构型IRMOF中节点Zn4O簇的转化。研究发现,通过在DEF中浸泡,Zn4O簇中的一个四配位Zn2+可以额外与一个DEF分子和一个水分子配位,形成六配位构型。上述结果证明,在常规Zn4O簇中四配位的Zn2+的确处于配位不饱和的状态,可作为潜在的Lewis酸性位点。随后的研究表明,该MOF可在较温和条件下(50 ℃和0.1 MPa CO2)实现多种环氧化物到环碳酸酯的高效转化(图式4)。上述报道通过对催化剂结构的精确表征,为MOFs中Zn4O簇具备Lewis酸催化活性提供了直接的结构证明。
(2)Cu2(COO)4簇
1999年,Williams等[37]报道了著名的HKUST-1,其分子式为[Cu3(BTC)2](BTC=1,3,5-苯基三羧酸)。该MOF的金属节点由双核Cu2+以及4组COO-组成,形成双核轮桨金属簇Cu2(COO)4。HKUST-1具有两种结构的孔道,一种为类似正八面体的小孔笼,另一种为三维正交的直孔道,且两种孔道之间相互贯穿。通常来说,该类型MOFs中Cu2(COO)4簇的两个Cu2+在轴向方向容易和两个弱配位的溶剂分子相连,使得Cu2+呈现四方锥配位构型。通过简单的加热活化可除去轴向配位溶剂分子,从而使Cu2+的不饱和配位点暴露在MOF孔洞当中,进而具有Lewis酸催化活性。
2017年,Zhao等[38]设计合成了一种富含酰胺基团的四羧酸配体,并利用其构建了一种基于Cu2(COO)4簇的新型MOF催化剂。单晶结构分析表明,Cu2(COO)4簇中Cu2+处于配位不饱和状态,具有潜在的Lewis酸催化活性。另一方面,基于其高孔隙率、富氮基团以及活性金属位点,该MOF可在室温条件下实现对CO2的高效捕获,吸附能力高于许多同类型MOFs。得益于这些特性,该催化剂在常温常压条件下可以催化一系列环氧化物高效转化为环状碳酸酯,且在同等反应条件下的催化活性明显高于HKUST-1。他们认为,较强的CO2吸附能力是该MOFs具备高催化活性的主要因素。值得一提的是,MOFs的孔洞择型作用使得分子尺寸较小的环氧化物具有高的催化活性,而大尺寸底物则受限于传质,活性非常低(图式5)。
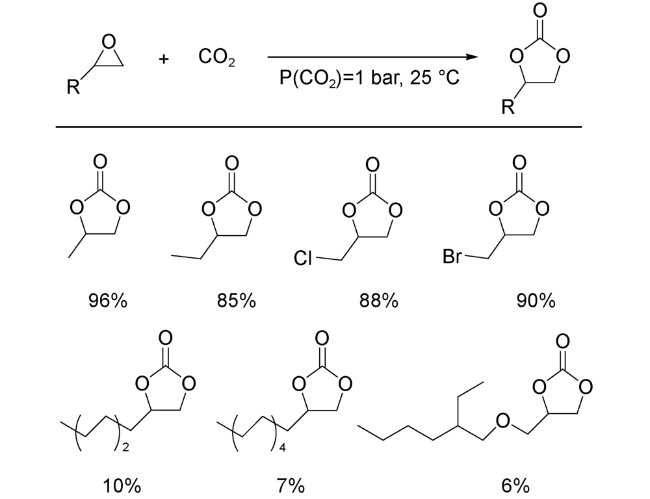
2018年,Ma等[39]发展了一例基于Cu2(COO)4簇的高化学稳定型MOFs材料JUC-1000,并利用其在常温常压条件下实现了环氧化物到环状碳酸酯的高效转化。在同等反应条件下,JUC-1000的催化活性显著高于HKUST-1和MOF-505等常见含有Cu2(COO)4簇结构的MOFs。他们提出,JUC-1000中配体上的—OH、—NH等极性基团可与配位不饱和的Cu2+协同作用,共同活化环氧化物,从而大幅度提高反应活性。
(3)M3FO(COO)6簇
MILs系列MOFs是由三价的Cr、Fe、Al等金属离子与BDC或BTC合成的,其中,最为典型的是Férey[40]报道的MIL-101(Cr) Cr3F(H2O)2O(BDC)3。该MOF的金属节点由3个Cr3+、6个COO-、一个μ3-O2-、一个F-以及两个H2O分子组成。其中,每个Cr3+分别与4个羧酸氧、一个μ3-O2-以及H2O分子或F-配位,呈现八面体的配位构型。MIL-101(Cr)中与Cr3+配位的水分子可以通过活化方式除去,使其暴露出不饱和配位点,从而具有Lewis酸催化活性。此外,该MOF含有两种内径为2.9和3.4 nm的空穴,分别通过直径约为0.9 nm的窗口连通成为三维孔道结构,其比表面积高达5900 m2/g。
2013年,Zalomaeva等[41]报道了MIL-101(Cr)在环氧化物转化环碳酸酯反应中的应用。研究表明,在MIL-101(Cr)与共催化剂TBAB的协同作用下,环氧丙烷和苯基环氧乙烷可以在较温和的条件下(25 ℃和0.8 MPa CO2)高效转化为相应的环状碳酸酯。然而,对于位阻较大的环己烷环氧化物,反应的活性则非常低。
2015年,Ma等[42]通过对BDC上引入的氨基进行后修饰,成功在MIL-101(Cr)框架内负载了季胺和季膦两类离子液体,得到了离子液体修饰的MIL-101(Cr)型双功能催化剂MIL-101-N(n-Bu)3Br 和MIL-101-P(n-Bu)3Br。得益于孔洞内固有的Lewis酸碱催化位点,该催化剂可在温和且无共催化剂存在的条件下,实现一系列环氧化物到环状碳酸酯的高效合成,且可循环多次利用(图式6)。
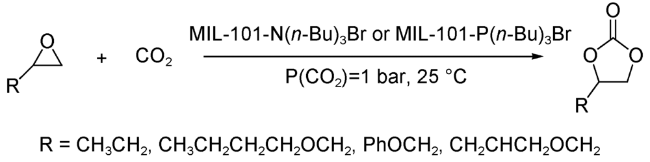
2018年,Jiang等[43]利用离子液体原位聚合策略,在MIL-101(Cr)孔洞内负载了聚合咪唑离子液体,从而实现了在MIL-101(Cr)中同时引入Lewis酸碱位点。通过聚合咪唑离子液体的引入,该MOF可在较温和、无共催化剂条件下实现一系列环氧化物高效高选择性地转化为环状碳酸酯,其催化活性明显高于未修饰的MIL-101(Cr)和聚合咪唑离子液体。他们认为,离子液体负载的MIL-101(Cr)中Lewis酸催化位点、Lewis碱催化位点以及其对CO2的强吸附性能共同促进了反应的高效进行。
(4)Zr6O8簇
UiO是一类著名的高稳定型MOFs系列[44]。该类MOFs一般由Zr6O8金属簇与直线型二羧酸配体构建而成,一般通式为Zr6O4(OH)4(COO)12。Zr6O8金属簇通常呈现12连接,其中,每个Zr4+分别与4个μ3-O2-和4个羧酸氧相连,呈八配位十二面体构型。通过高温活化,UiO型MOFs的节点可脱去两个水分子变为Zr6O6,同时Zr4+变为七配位不饱和状态,具备潜在的Lewis酸催化活性。另一方面,通过调控合成条件,可以得到Zr6O8金属簇的配体连接数分别为10、8、6的不同MOFs,而在这类MOFs中,Zr6O8金属簇具备更多的Zr4+开放位点,同样可以用于Lewis酸催化反应。
2013年,Ahn等[45]系统比较了UiO-66、UiO-66-NH2、Mg-MOF-74、MIL-101、HKUST-1、ZIF-8、IRMOF-3、MOF-5等不同类型MOFs在环氧化物合成环状碳酸酯反应中的催化活性。首先,他们通过NH3-和CO2-TPD测试,表征了以上不同类型MOFs的Lewis酸碱性。结果表明,大部分MOFs只单独具备Lewis酸性或Lewis碱性,而UiO-66-NH2则同时具备适宜的Lewis酸性和Lewis碱性。随后的催化结果表明,UiO-66-NH2可高效地催化苯基环氧化物转化为相应环状碳酸酯,反应在1 h内可达70%收率,4 h内反应完全,效率远高于其他几种MOFs催化剂。上述研究结果说明,MOFs内Lewis酸碱的共同作用对环氧化物与CO2偶联合成环状碳酸酯的反应是必不可少的。
2016年,Su等[46]发展了一种基于UiO-66-NH2纳米颗粒的金属有机凝胶材料。对比常规的UiO-66-NH2,金属有机凝胶材料在同等条件下的CO2吸附能力增加了38%。另一方面,由于金属有机凝胶含有较多的缺陷位点,因而具备更多的Zr4+开放位点。得益于上述两方面的优势,金属有机凝胶材料可在相对温和的条件下催化一系列环氧化物转化为环碳酸酯,其催化效率明显高于UiO-66-NH2。
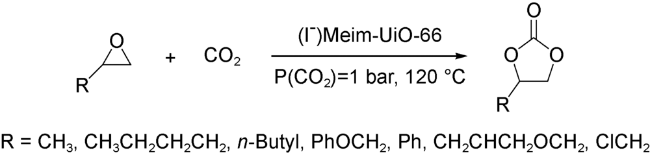
2017年,Cao等[47]通过后修饰的手段,设计合成了一种咪唑碘盐修饰的UiO-66型催化材料。他们首先利用咪唑修饰的对苯二羧酸构建了Im-UiO-66,并在此基础上使用CH3I对Im-UiO-66进行后合成修饰得到了(I-)Meim-UiO-66。在催化反应研究中,他们发现在同等反应条件下,UiO-66、UiO-66-NH2以及Im-H2BDC和ZrCl4的混合物都无法有效催化反应的进行,而(I-)Meim-UiO-66中Zr6O8中的Zr-OH/Zr-OH2酸性位点和配体上的I-碱性位点则能高效地促进反应的发生(图式7)。同年,Dong等[48]利用类似策略构建了一例咪唑溴盐修饰的UiO-67型催化材料。同样,该催化剂可以在无共催化剂存在的条件下,催化一系列环氧化物高效转化为环状碳酸酯。
(5)其他金属簇
MOF-74由二价金属离子(Cu、Zn、Co、Mn、Mg、Cd等)与配体2, 5-二羟基对苯二甲酸自组装构成,形成一维六方孔道、三维类蜂窝状的空间网络结构,孔径约1.1~1.2 nm。节点金属簇[M3O3(COO)3]中每个金属离子配位数为6,其中5个氧原子来自配体上的羧基和羟基,另1个配位原子来源于溶剂或水分子,通过真空加热可以得到位于孔道内表面的不饱和金属位点[49]。2012年,Ahn等[50]研究了Mg-MOF-74在298 K时的CO2吸附能力,并采用循环吸附-解吸实验证实了Mg-MOF-74对CO2的完全可逆吸附和高稳定性。在相对温和的条件下(100 ℃和2 Mpa CO2),活化后的Mg-MOF-74在CO2和苯乙烯环氧化物反应中表现出良好的催化性能(产率95%)。
Han等[53]通过原位配体反应合成了一例链状金属簇[In(μ2-OH)In]基MOF。他们通过实验证明,MOF中开放的金属In位点作为Lewis酸中心,而μ2-OH基团作为Brønsted酸中心,原位配位反应嫁接的哌嗪不仅有助于框架的稳定,还有利于CO2的选择性吸附。该MOF作为Lewis/Brønsted酸性双功能非均相催化剂,可在温和的条件下(室温和1 atm CO2)高效催化环氧化物与CO2偶联合成环状碳酸酯。此外,该催化剂具有良好的可回收性,在5次循环反应后其活性没有下降。2017年,他们又构建了一例稀土金属簇[Yb5(μ3-OH)6]基MOF[54]。由于配体中Brønsted酸性-COOH基团和Lewis酸性Yb(Ⅲ)位点的协同作用,该MOF具有较强的CO2亲合力,并在环氧化物与CO2偶联合成环状碳酸酯反应中表现出优异的催化活性。
基于稀土离子较高的配位数和较强的Lewis酸催化性能,2017年,Zhao等[55]报道了两例基于[Tb4(μ3-OH)4]簇的MOFs。金属簇中每一个Tb3+的配位数为7,属于不饱和配位,具有Lewis酸催化位点,因而在环氧化物与CO2偶联合成环状碳酸酯反应中表现出较好的催化活性。
理论上,任何节点(单金属离子或金属簇)处含有不饱和配位点的MOFs都具有潜在的Lewis酸催化活性。然而,需要指出的是,MOFs节点作为催化位点对其框架稳定性有一定的影响。决定MOFs结构是否稳定在于节点与配体间的配位键强度,而节点对底物的活化一定程度上会干扰MOFs框架配位键。对于一些节点与配体间配位键强度较弱的MOFs,其框架结构容易在催化反应过程中坍塌,降低其重复使用性能,这也是直接利用MOFs节点催化的一大弊端。尽管如此,MOFs节点的多样性仍使得其成为MOFs非均相催化领域中最常见的一种方式。
2.3 MOFs功能有机配体Lewis酸催化位点
MOFs中的功能有机配体同样可以起到Lewis酸催化的作用,而且对MOFs框架结构没有影响,其中,金属卟啉配体和金属salen配体是目前研究最为广泛的两种功能配体。
2013年,Ren等[56]设计合成了一种新型salen-Ni金属有机配体,并利用其构建了一例含有一维孔洞的三维Cd-MOF。单晶结构分析表明,Ni2+分别与salen配体上的2个氮原子和2个氧原子配位,呈现四配位平面正方形构型,具有潜在的Lewis酸催化性能。催化结果表明,Cd-MOF/TBAB双催化剂体系,可在较温和的条件下促使不同的环氧化物高效转化为环状碳酸酯。然而,单独使用salen-Ni配体或Cd(bpdc)n都不能有效地促进反应的进行。因此,他们认为Cd-MOF中salen配体上Ni2+和金属节点Cd2+的协同Lewis酸催化、以及MOF孔道对CO2的吸附作用共同促进了反应的高效进行。
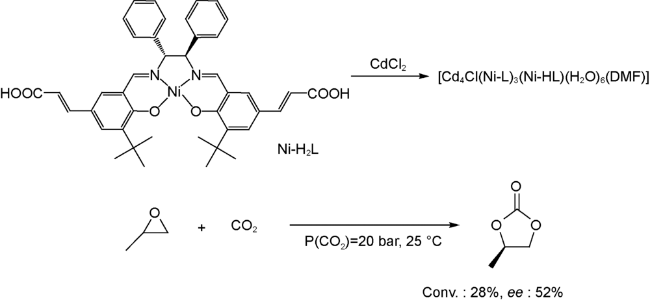
同年,他们又报道了另一例基于手性salen-Ni配体的三维手性Cd-MOF,并利用该MOF首次实现了环氧化物到环状碳酸酯的不对称转化[57]。在该催化体系中,salen配体中Ni2+作为Lewis酸催化位点,而共催化剂TBAB作为Lewis碱,同时,salen配体上的二苯基团则起到手性诱导的作用。在三者的共同作用下,该MOF可在室温、2 MPa CO2压力下催化环氧丙烷以28%的转化率和52%的ee值得到目标产物(图式8)。尽管反应转化率和ee值不高,但是该报道首次把手性MOF应用于环氧化物到环状碳酸酯的不对称转化当中,为后期相关研究奠定了基础。
2017年,Ren等[58]又设计合成了一种吡啶型salen-Ni配体,并利用其构建了一例具有高水、热及化学稳定性的三维手性MOF。单晶结构分析表明,该MOF具备罕见的八重穿插结构。同时,salen配体上的二苯基伸向MOF孔洞内,形成了疏水孔道结构。基于其结构特性,该MOF可在高温(>400 ℃),酸碱及氧化性溶液(包括1 M HCl溶液、饱和NaOH溶液、30 wt% H2O2溶液以及70 wt% TBHP溶液)以及水蒸气中保持结构稳定。另一方面,salen-Ni配体中的Ni2+呈四配位不饱和状态,且其不饱和配位点正好朝向MOF孔洞之内,形成了适合Lewis酸催化微环境。结合以上特点,该MOF可催化环氧化物与模拟工业用CO2(含少量SO2和H2O)高效转化为环状碳酸酯,且催化剂可在此严苛条件下重复使用3次而保持活性不变。
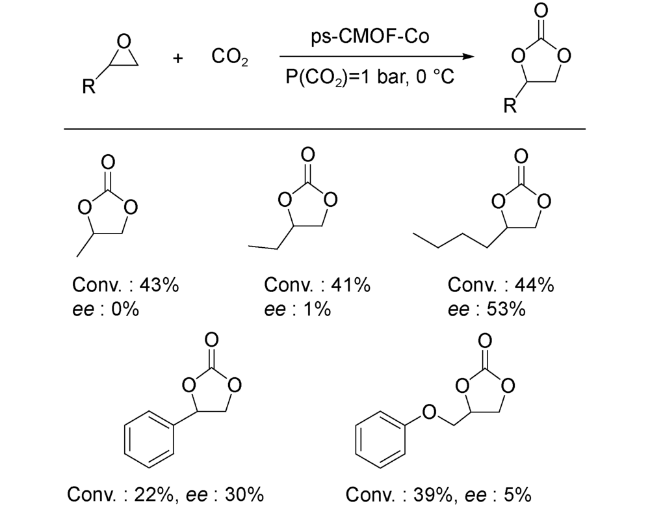
2018年,Ren等[59]巧妙地结合金属卟啉和金属salen配体,首次构建了卟啉-salen基手性ps-CMOF,并通过后修饰的手段调控ps-CMOF中卟啉中心金属和节点金属,合成得到了一系列具有不同金属位点的ps-CMOFs。CO2吸附等温线数据表明,Co修饰的ps-CMOF可在室温下高效吸附CO2,优于同构型的其他MOFs。同时,单晶结构分析表明,金属salen配体的二苯基团位于金属卟啉中心Co2+正上方,形成良好的手性催化微环境。因此,他们将ps-CMOF(Co)应用到环氧化物转化为环状碳酸酯的不对称反应中。催化结果显示,该MOF可在温和条件下以最高44%转化率、53%的ee值得到目标产物(图式9)。随后,他们通过控制实验证实,在ps-CMOF(Co)中,金属卟啉中心的Co2+起到Lewis酸活化的作用,而金属salen配体的手性二苯基团起到手性诱导作用,二者之间的协同作用共同促使了反应的进行。
同年,Cao等[60]通过连续后合成离子化-金属化的策略,构建了含有Zn-四-(4-羧苯基)卟吩(Zn-TCPP)、咪唑溴盐的新型Zr基MOF催化材料ZnTCPP⊂(Br-)Etim-UiO-66。在该催化体系中,ZnTCPP⊂(Br-)Etim-UiO-66中的卟啉配体中心Zn2+起到Lewis酸催化作用,而咪唑配体上的Br-为Lewis碱,两者之间协同作用可在无共催化剂条件下催化一系列环氧化物转化为环碳酸酯。
2019年,He等[61]设计合成了一系列结合金属卟啉配体和五氟氧铌(NbOF5)2-的新型柱层结构多元MOFs(Cu-Nb-Ni、Cu-Nb-Fe、Cu-Nb-Zn)。MOFs结构分析显示,该框架内含有卟啉中心金属、节点Cu2+以及(NbOF5)2-等多种有效位点,可大幅提高框架对于CO2气体的亲合力。CO2吸附等温线数据表明,当卟啉中心的金属为Ni2+时,MOF对CO2的吸附能力最强。得益于其多金属开放位点和CO2亲合力,Cu-Nb-Ni可在常温常压下实现一系列环氧化物高效转化为环状碳酸酯,且循环利用3次以上而未失去活性。
2.4 MOFs双催化位点
除了金属节点和金属有机配体,通过结构的合理设计将有可能在MOFs框架内引入多种金属活性位点,协同催化有机反应的发生。MOFs材料高度可调控的特点赋予了其结构设计的多样性。
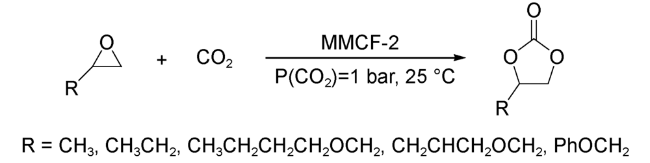
2014年,Ma等[62]通过晶体工程策略,构建了一例与MOF-505具有相同nbo拓扑结构的新型MOF材料MMCF-2。结构分析表明,MOF-505和MMCF-2都是由12组Cu2(COO)4金属簇充当顶点所构成的十四面体笼结构。与MOF-505不同的是,MMCF-2中氮杂大环四羧酸配体tactmb(1,4,7,10-tetrazazcyclododecane-N,N',N″,N‴-tetra-p-methyl-benzoicacid)中心可以和一个Cu2+配位,充当额外的催化活性位点。催化性能研究表明,MMCF-2可以在常温常压条件下催化多种环氧化物高效转化为环状碳酸酯(图式10),同时,其催化活性明显高于MOF-505、HKUST以及Cu-tactmb。上述结果说明,MMCF-2催化合成环碳酸酯的过程中,Cu2(COO)4金属簇和配体中心的Cu2+充当双金属催化位点,协同催化促进反应的进行。
2018年,Morris等[63]报道了基于金属Cyclam配体的两例新型Zr基MOFs VPI-100(Cu)和VPI-100(Ni)。单晶结构显示,VPI-100由Zr6金属簇与8个金属Cyclam配体相连接,因此,Zr6金属簇中含有开放的金属位点可用于Lewis酸活化。同时,VPI-100(Cu)和VPI-100(Ni)中的金属Cu2+和Ni2+分别与Cyclam环中的4个N原子配位,其轴向方向仍有空配位点,同样可用于Lewis酸活化。基于以上结构特点,他们把VPI-100(Cu)和VPI-100(Ni)应用到环氧化物合成环碳酸酯的反应中。结果显示,VPI-100(Cu)和VPI-100(Ni)可高效催化反应发生,且在已报道的Zr基MOFs中,其TOF值最高。他们认为,节点Zr4+与金属Cyclam中的Cu2+或Ni2+之间的协同Lewis酸活化是促进该反应高效进行的关键。
2018年,Fischer等[64]系统考察了在Zr6金属簇连接数分别为12、8、6的MOF-525, PCN-222和PCN-224中,Zr6金属簇Lewis酸单催化位点以及引入第二Lewis酸协同催化位点对环状碳酸酯合成反应的影响(图式11)。催化结果表明,对于Zr6金属簇作为单催化位点,其连接数越低,金属开放位点越多,反应活性越高。通过后修饰在卟啉配体中心引入第二金属Mn2+之后,三种MOFs的反应活性都有所提升,其中,连接数分别为12、8的MOF-525和PCN-222的活性提升更为明显。他们认为,配体连接数更多的MOF可以负载更多的Mn2+,因此,第二金属的引入对反应活性的影响也越明显。
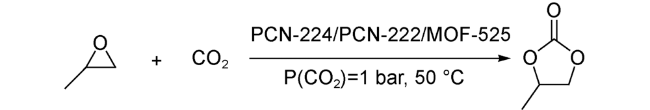
3 MOFs催化CO2与末端炔的羧基化反应
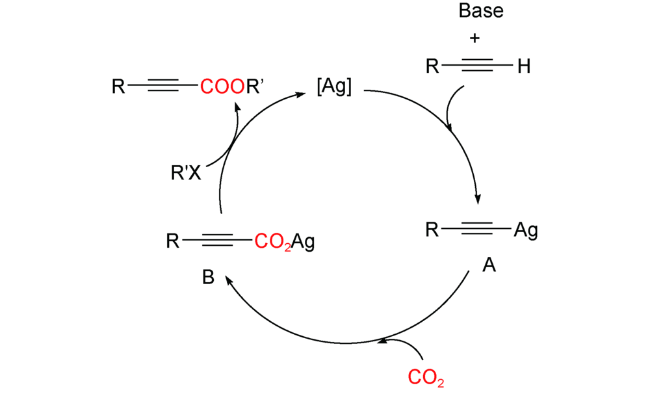
2015年,Cheng等[68]采用液体浸渍-还原法,在MIL-101(Cr)上负载银纳米粒子得到了一系列Ag@MIL-101复合材料,其中,Ag纳米粒子的平均尺寸为1.4 nm, 负载量为1.66%~6.97%(质量比)。以Ag@MIL-101为催化剂,末端炔和CO2(1 atm)为原料,可在50 ℃下,DMF中合成炔丙酸衍生物(图式13),而且底物适用性高,含有吸电子、给电子的芳香末端炔和杂环炔均可发生反应,产率高达98%。此外,催化剂可通过离心回收,至少使用5次催化活性并未降低。他们认为Ag@MIL-101的高比表面积和多孔性在该催化反应中发挥了重要作用:(1) 该材料能有效地捕获CO2,提高框架孔内催化中心附近的CO2浓度,促进反应的进行。(2) MIL-101的框架和孔作为“微反应器”,为CO2与末端炔烃反应提供了良好的微环境。由于配位不饱和Cr3+的存在,框架的Lewis酸非常强,可以优先吸附芳香族底物,从而提高催化剂的活性。(3) MIL-101的高稳定结构使复合材料更加稳定,并具有良好的再生性能。

采用类似策略,2017年,Ma等[69]使用更加环保的MIL-100(Fe)和UiO-66来进行修饰。与Ag@MIL-101相比,Ag@MIL-100(Fe)具有相似的催化活性、稳定性和可重复使用性,但对环境更友好,易于合成。另一方面,Ag@UiO-66在捕获CO2和掺杂Ag纳米颗粒方面更为高效和经济,但由于其孔道狭窄,其可重复利用性较低。
2018年,Natarajan等[70]在溶剂热条件下,以1,2-二氨基-3,6-双(4-羧基苯基)苯和硝酸锌为原料,合成了一种三重互穿三维卟啉基MOF,并通过液体浸渍-还原法得到了负载Ag纳米粒子的MOF复合物。在温和条件下(60 ℃和1atm CO2),以其作为异相催化剂,可高效催化末端炔烃羧基化转化为炔丙酸酯,并具有良好的稳定性和可回收性。此外,他们研究了不同Ag含量(3.0%、7.8%和15.0%)样品的催化性能。结果发现,3.0%负载量的样品中Ag纳米粒子的粒径约为3.0 nm,且分布均匀;7.8%负载量的样品中Ag纳米粒子的粒径约为3.8 nm;而较高负载量(15.0%)则会导致银纳米粒子的团聚。在相同实验条件下,以苯乙炔为底物,7.8%负载量样品显示出最优的催化性能,说明Ag纳米粒子的尺寸对催化活性具有重要影响。
4 MOFs催化CO2与炔丙胺/醇的环化反应
2018年,Duan等[71]利用层扩散法,采用设计合成的含硫脲配位点的三脚架型配体与AgBF4构建了一例新颖的银簇基MOFs。研究发现,以该MOFs为催化剂,在室温常压下炔丙胺衍生物和CO2可以发生环化反应生成α-亚烷基环碳酰酯产物(图式14)。控制实验表明简单银盐(AgOTf、Ag2O、AgNO3、AgI、AgOAc、AgCF3COO和AgBF4)在标准条件下并不能有效催化该反应的进行,说明MOFs的框架结构、Ag-π(C≡C)活化和氢键作用共同促进该反应。不足的是,该反应底物仅限于末端炔烃,苯基取代的炔丙胺由于其空间位阻大而不发生反应。
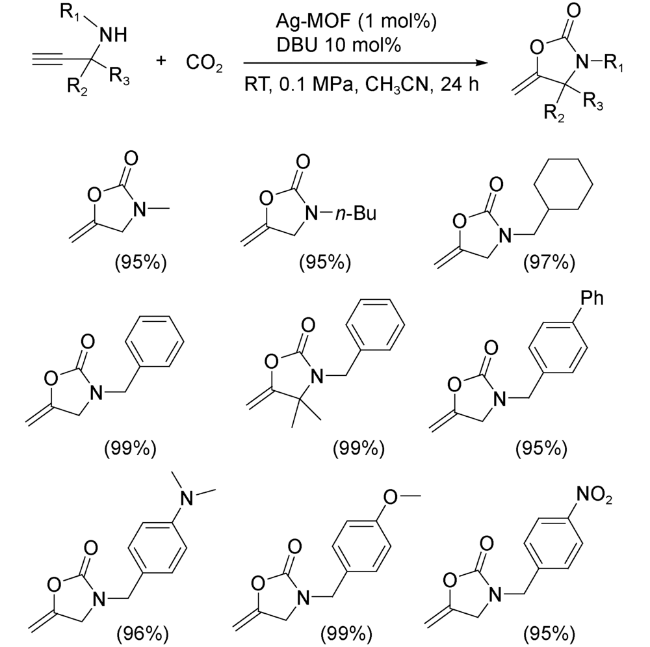
同年,Fei等[72]用Cu(NO3)2·3H2O、4,4'-联吡啶和1,2,4,5-四乙基磺酸苯,采用溶剂热法合成磺酸基MOFs,并进一步利用MOFs中两个未配位磺酸基团络合Ag+。研究发现,以该负载Ag+的MOFs为催化剂,在常温常压下炔丙醇衍生物和CO2可以发生环化反应生成α-亚烷基环碳酸酯产物(图式15)。其中,大多数底物的产率几乎是定量的。同时,内丙炔醇只能以25%的收率得到相应的碳酸盐,其原因可能是其空间位阻大难以扩散到MOFs孔腔内,以及Ag+位点对内炔烃π活化不足。
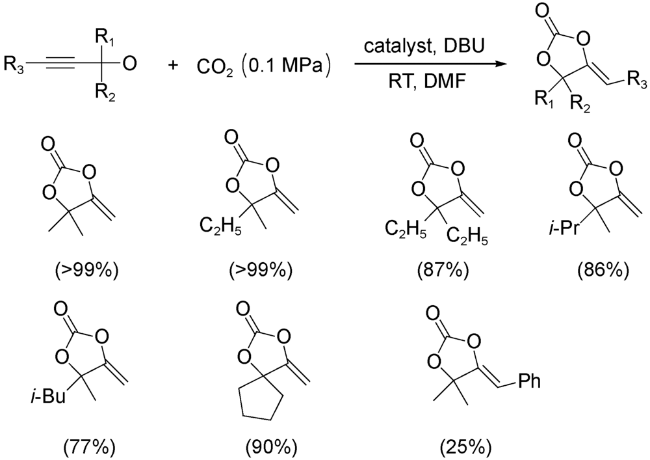
2019年,Zhao等[73]以CuI、In(NO3)3和5-氨基烟酸为原料,在溶剂热条件下合成了一例具有开放纳米孔道的异金属MOFs(图式16)。该MOFs具有较高的溶剂稳定性和酸碱稳定性。实验结果表明,该MOFs在较温和条件下能有效催化CO2与各种末端炔丙醇发生羧基化环化反应,TON值高达14 400。他们通过控制实验证明MOFs纳米通道中[Cu4I4]团簇为关键的催化位点,In3+是共催化剂,可以进一步促进环化反应。重要的是,这是第一个无贵金属MOF直接催化炔丙醇与CO2的环加成反应。
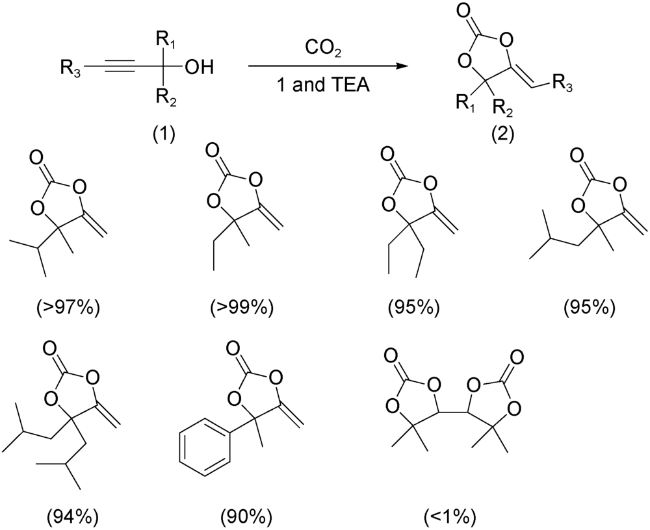
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
5 结论
MOFs催化位点的可设计性赋予了其在化学固定CO2方面新的发展契机,并已取得了重要的进展,尤其是MOFs较高的比表面积以及孔道/孔腔的可修饰性为CO2的吸附和活化提供丰富的探索空间。CO2的高效活化与化学固定研究不仅需要新颖,更需要追求其最后的实用价值。因此,高效、高稳定(对水、酸、碱、空气/湿气、高温的耐受性)、低成本MOFs的设计构建,以及利用MOFs发展更加绿色、经济、可持续的CO2化学固定方法学将是该领域未来的研究热点。