



05-0649-21_pic.jpg)
1 引言
不论是“老药”胰岛素、放线菌D,还是近期的热点药物司美格鲁肽、替西帕肽,肽类药物一直是新药研发的热点领域。肽类药物所展示出来的抗肿瘤、抗耐药菌、抗血管生成和抗凝血等广泛的生物活性,以及低毒性、高特异性和低免疫原性等药效学特点,极大促进了现代新药的研发[1]。目前,临床上广泛使用的小分子药物通常只能靶向具有明确结合口袋的靶点。同时,由于小分子药物固有的小尺寸,使得与靶点结合时,平坦、宽广的靶标表面不能被完全占据[2]。据统计[3],在体内众多与疾病相关的蛋白质靶点中,小分子药物只能与体内约10%的靶点有靶向性和亲和力。特别是对于蛋白质-蛋白质相互作用的靶点(PPIs),小分子药物很难与其结合,是“不可成药的”,而肽类药物可以很好地解决这些问题。肽类药物可以分成2大类——线性肽和环肽。最早开发的肽类药物大部分是天然线性多肽,例如胰岛素、降钙素、抗利尿激素等,这些肽类药物实际上是“代替疗法”,用于补充内源性肽水平不足或者缺乏[4]。线性肽具有一些局限性,例如物理化学性质不稳定、易被体内的酶水解、口服生物利用度低等[5]。基于这些缺点,研究人员开始使用药物化学相关技术,对肽类药物进行化学修饰,由此开发出来一系列环肽化合物。环肽比抗体、蛋白质类药物分子质量小,部分环肽可以透过细胞膜[6]。环肽结构中酶切位点不易被外肽酶接触到,加之骨架柔韧性较差,阻碍了酶活性位点的契合[7],因此对外肽酶的稳定性提高,在体内的作用时间延长。此外,环肽结构的刚性可以降低吉布斯自由能的熵值,增强与靶点的结合能力[8]。然而,随着肽环的增大,环肽构象上开始变得灵活,容易被水解酶破坏,并且构象的改变降低了与靶点的亲和力,甚至出现“脱靶”现象。最佳方法是将单环转变为双环,这样,环的尺寸减小了一半,刚性增加,化学和水解稳定性可进一步提高[9]。
2 双环肽的介绍
2.1 结构特点
双环肽,顾名思义是结构中含有2个环的肽,目前对双环肽结构的分类尚未有明确的定义,本文将常见类型的双环肽进行了结构划分,具体分为如下4类(图1)。
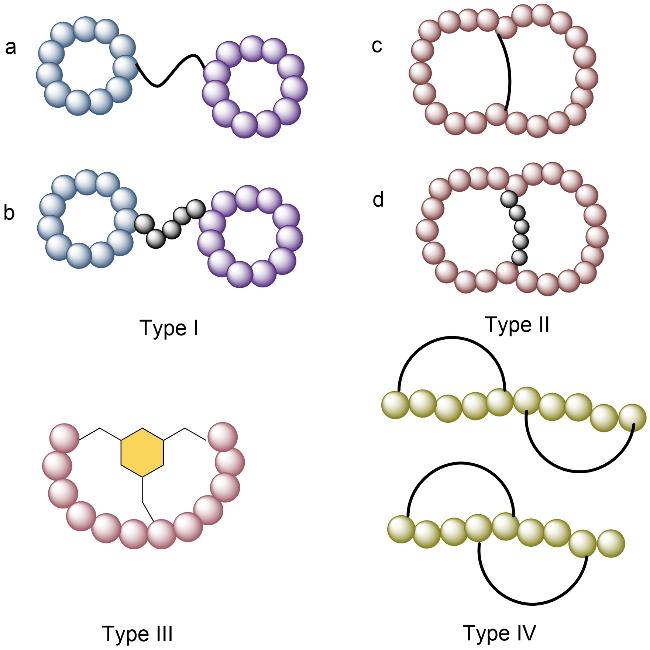
(1)类型I,2个单环肽相连。其中连接体可以是线性肽链,也可以是小分子连接体,但以小分子连接体为多,例如放线菌素D。
(2)类型II,在首尾相连的大环肽内部桥连。由此得到2个肽环之间存在共用部分的双环肽,同样,桥连部分可以是肽链,也可以是小分子刚性连接体。类型II的双环肽在自然界中居多,例如α-Amanitin、theonellamide F等,也可以通过两步环化进行化学合成。
(3)类型III,手柄结构的双环肽。小分子化合物被用作连接肽链的骨架,形成类似“方向盘”结构。小分子有多种选择,双环肽的构建相对快捷、方便,更适合大量快速合成,被广泛应用于双环肽库的构建,尤其是生物法构建双环肽库。
(4)类型IV,钉书肽(stapled peptides)。钉书肽是指在线性肽主链上引入可连接的基团,通过烯烃复分解、点击化学等方法环化构建而成。订书肽具有α-螺旋程度高、细胞膜通透性强、难以被蛋白酶水解,以及在生物体内半衰期长等优点[12]。
2.2 天然双环肽
以天然产物为先导化合物进行结构修饰是新药研发的重要手段,天然双环肽在自然界有着广泛的分布,具有各种各样的生物活性,不断激励着双环肽药物的研发。
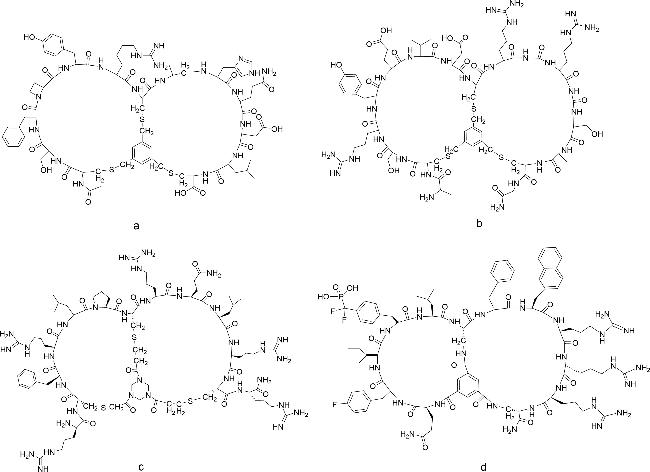
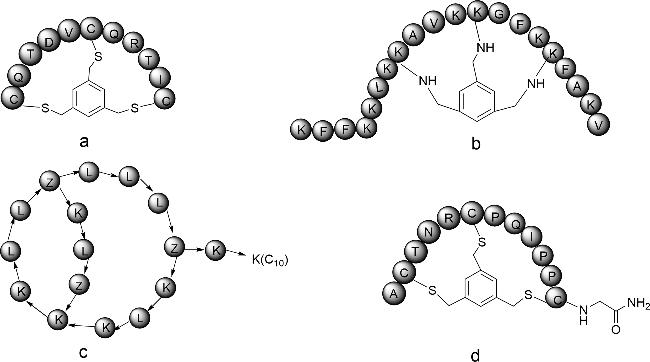
放线菌素D是一种抗肿瘤多肽抗生素(图式1a),是治疗癌症的老药,于20世纪60年代被批准用于肿瘤治疗[13]。2016年,Liu等[14]将放线菌素D与RG1718(靶向间皮素的免疫毒素)联合应用,发现两者产生协同作用,增强了在人体内的抗肿瘤活性。另一个已经上市的天然双环肽是罗米地辛,从日本土壤样品中分离出来的紫色杆菌中获得,是一种组蛋白去乙酰化酶抑制剂,于2009年被批准用于治疗皮肤T细胞淋巴瘤。其结构中含有1种天然氨基酸、3种非天然氨基酸,以及1种非氨基酸构建块(图式1b)[15]。α-Amanitin是从死帽菌中分离出的剧毒双环八肽(图式1c),能够高选择性抑制RNA聚合酶II,对真核细胞产生毒性,最终导致细胞凋亡[16]。2018年,Matinkhoo等[17]对α-Amanitin完成了全合成,克服了6-羟基-色氨酸亚砜桥,(2S,3R,4R)4,5-二羟基异亮氨酸的对映选择性合成和非对映选择性亚砜化的关键挑战。α-Amanitin现作为抗体-药物偶联物(ADC)在癌症治疗中的研究越来越广泛。例如,将抗EpCAM单抗与α-Amanitin偶联嵌合,得到抗体-药物偶联物chiHEA125-Ama [18],可有效根除实验性胰腺癌,且具有较低的全身毒性风险,因此可能成为胰腺癌和过表达EpCAM的新型抗癌药物。2004年,Potterat等[19]从链霉菌培养液中分离到一个新的双环19肽BI-32169(图式1d),对人胰高血糖素受体具有强抑制活性。Kim等[20]从萨夫芽孢杆菌KCTC 12796BP培养液中分离得到了6种化合物,其中4种为双环肽(图式1e),2种为单环肽。化合物的生物活性测试结果表明,只有双环肽具有抗过敏的活性,而单环肽不具有活性,此发现为抗过敏药物的研发提供了一个新的方向。Karim等[21]从日本海深海悬浮物中分离的链霉菌中发现了2个双环肽,Nyuzenamides A和B(图式1f Nyuzenamide A、B),2种化合物均存在抗真菌活性和细胞毒性。随后,An等[22]在相同来源中发现了另一种双环肽——Nyuzenamide C(1)(图式1f)。通过进一步的生物活性测试,研究人员发现Nyuzenamide C(1)在人脐静脉内皮细胞中显示抗血管生成活性。
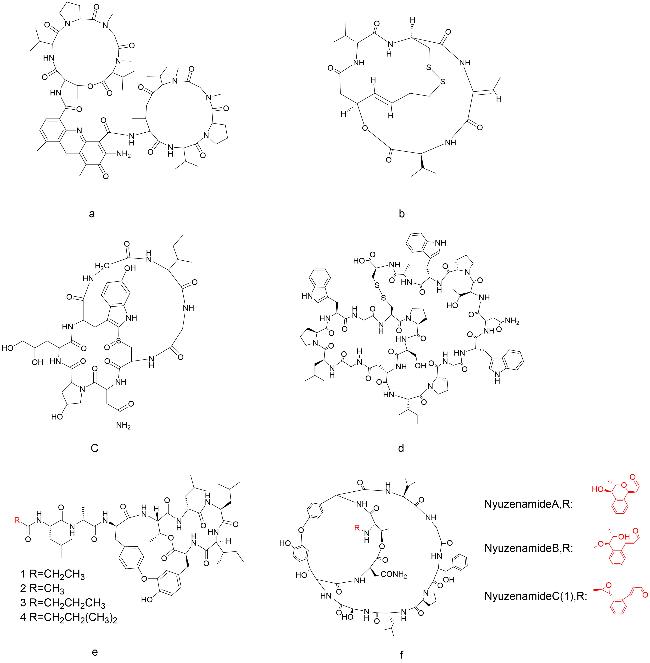
除了微生物来源的双环肽以外,许多植物中也分离出了双环肽。1989年,报道了一种由组氨酸和丙氨酸残基桥接的双环肽theonellamide F[23](图式2a),该肽是从海洋植物海绵的次级代谢产物中分离而来的,是一种具有抗真菌活性和细胞毒性的双环肽。随后,对该海绵提取液的抗真菌部分进一步分离纯化,得到5个相关肽,经过活性实验,发现均存在不同程度的抗真菌活性和细胞毒性[24]。向日葵胰蛋白酶抑制剂-1(SFTI-1)(图式2b),从葵花籽中提取得到,是目前自然界中发现的最小的二硫桥环肽之一[25],也是最小和最有效的BBI(Bowman-Birk inhibitor)之一[26]。其结构被1个二硫桥分成2个环:1个功能性胰蛋白酶抑制环(TI)和1个非功能性二级环(SL),后者可以被生物活性肽取代[27]。SFTI-1还具有抗菌活性[26]、促血管生成活性等[27]。从Rubia cordifolia L.和Rubia argyi(H. lsamv.)的根中分离出来的双环六肽—RA-VII(图式2c),研究表明,该双环肽通过与真核核糖体相互作用,抑制蛋白质的合成,从而产生强大的抗肿瘤活性[28]。青葙子为苋科植物Celosia argentea L.的干燥成熟种子,是中医上常用的一味中药之一,常用于治疗一些眼部疾病。从青箱子中提取出的一种双环八肽——Moroidin[29](图式2d),能够通过抑制微管蛋白聚合,而产生抗肿瘤活性,其对微管蛋白聚合的抑制活性高于秋水仙碱[30]。
3 双环肽的合成
含有二硫键的肽在自然界中广泛存在,二硫键赋予了良好的热稳定性和水解酶稳定性,以及结构多样性[32]。受此启发,在构建双环肽的方法中,利用半胱氨酸的巯基形成二硫键的方法尤为突出,是人工合成双环肽的常用策略。2001年,Sun等[33]报道了一种合成两亲性双环肽的有效的方法。线性肽分子中包括N端的半胱氨酸(Cys)、C端的硫酯结构以及内部的Cys。第1步环合是N端的Cys和C端的硫酯形成分子内硫酯的结构,然后经过自发的S,N-酰基转移,形成稳定的内酰胺。在pH值为5~6的缓冲水溶液中,DMSO介导第2步环化,末端的Cys与内部Cys形成树脂外的分子内二硫键,构建了第2个环(图式4a)。通过此方法合成了4个双环肽,并成功构建了由内酰胺和分子内二硫键约束的14个氨基酸组成的双环肽库。这些双环肽均表现出两亲肽所期望的性质,例如,优异的水溶性和膜亲和力。二硫键介导的双环肽合成方法已经非常成熟,近期,Rayala等[34]优化了含2个二硫桥的固相合成,并且成功合成了双环肽OL-CTOP。OL-CTOP具有鼻-脑递送能力,经过生物活性测定发现,经鼻给药OL-CTOP能够剂量依赖性地拮抗吗啡的镇痛作用,并防止吗啡引起的呼吸抑制,这为临床上研发吗啡急性中毒的解救剂开辟了新的思路和途径。
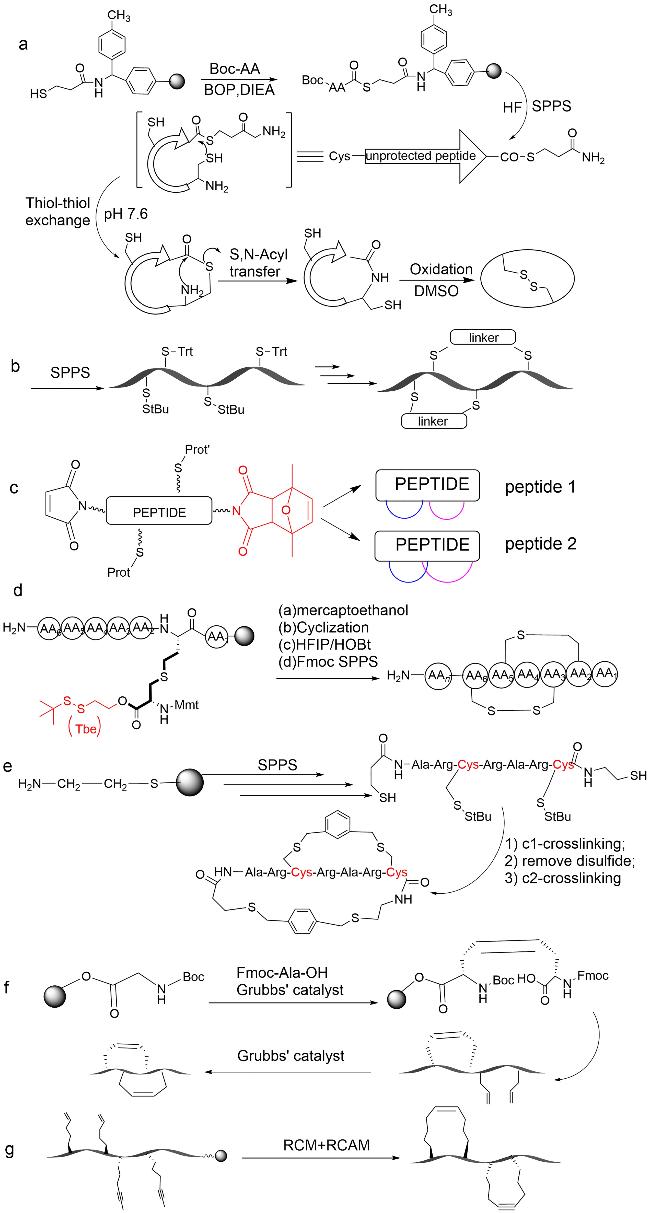
图式4 双环肽合成方法示例(a:通过树脂上分子内硫酯连接和树脂外二甲基亚砜介导的二硫键形成的合成方法;b:Trt-StBu策略用于硫醚双环肽的合成;c:Michael加成法合成双环肽;d:3个正交保护基团(Fmoc、Mtt和Tbe)的二氨基二酸法;e:头尾相连构建双环肽;f:烯烃复分解法;g:烯烃、炔烃复分解法相结合)Scheme 4 Summary of bicyclic peptide synthesis methods(a: Synthesis via intramolecular thioester linkage on the resin and dimethyl sulfoxide mediated disulfide formation outside the resin; b: Trt-StBu strategy for the synthesis of thioether bicyclic peptides; c: Michael addition for the synthesis of bicyclic peptides; d: diaminodicarboxylic acid method of three orthogonal protecting groups(Fmoc,Mtt,and Tbe); e: head-to-tail linkage to construct a bicyclic peptide; and f: alkyne reductomerisation method; g: combination of olefinic and alkynic complexation.) |
然而,在体内还原条件下(例如,谷胱甘肽),以及存在二硫键异构酶时,二硫键极易发生结构重排而导致生物活性减弱[35]。为了克服这个问题,人们多采用更稳定的结构来代替二硫桥,例如,硫醚、酰胺、烯烃、三唑和碳氢化合物桥等。替换二硫化物通常可以提高肽的稳定性,并保持其结构和活性。硫醚键由于与二硫键具有相似的结构参数,是最常用的二硫键代替物,合成方法主要有硫醇双烷基化反应、Michael加成、二氨基二酸的固相肽合成等方法。2021年,Zhu等[36]报道了一种新的硫醚键合双环肽的方法(图式4b)。该方法通过使用Trt保护和 S-StBu 保护的Cys来制备双环肽,在完成所有指定氨基酸的组装后,使用88%TFA裂解树脂,从树脂释放肽片段的同时脱除Trt保护基,使一对巯基裸露,通过硫醇双烷基化反应生成单环肽。随后,使用TCEP原位脱除StBu保护基从而使得另一对巯基裸露,再通过硫醇双烷基化实现双环终产物的合成。该方法不仅避免了传统方法去保护的烦琐步骤,而且降低了成本,更重要的是,该方法中2个硫醚键的形成是在溶液中进行的,用高效液相色谱法可以很容易地进行跟踪。
由于硫醚键的代谢稳定性有限,采用烯烃和烷烃桥来代替硫醚键可进一步提高代谢稳定性。Ghalit等[40]采用烯烃复分解(RCM)(图式4f)合成了一系列抗生素Nisin的双环模拟物,用烯烃和烷烃桥来代替硫醚键,模拟天然抗生素Nisin硫醚共价约束。所得到的Nisin类似物与天然Nisin进行对比,虽然对靶点的亲和力降低,但仍然具有一定的活性,可作为设计基于Nisin的新型肽类抗生素的第一代先导物。Cromm等[41]联合应用正交闭环炔与烯烃复合法(RCAM/RCM),成功地将小GTPase Rab8的单环肽抑制剂进化为双环配体(图式4g)。该双环肽显示了迄今为止报道的对Rab GTPase的最高亲和力。通过这2种方法,可以得到“钉接肽”,提高肽的α螺旋程度,从而提高膜通透性和代谢稳定性。
除了上述方法外,合成双环肽的一种简单方法是用不同的连接剂连接2个单环肽。例如,Mendive等[42]通过分子内钯催化(Pd(OAc)2)的C-H活化过程,制备色氨酸和苯丙氨酸或酪氨酸残基之间共价键连接的独特约束肽。钯催化偶联是合成环肽的有力方法,除此之外,还可以通过在单环肽序列中插入可偶联的基团来构建双环肽,例如点击化学法[43]。在细胞穿透肽(CPPs)中,环肽由于具有较高血清稳定性而具有更大的优势,研究表明,带正电的精氨酸和疏水色氨酸由于其与磷脂膜的相互作用特性,更大程度的提高了细胞穿透能力[44]。Oh等[45]以单环肽为基础,合成了2种由色氨酸和精氨酸残基组成的双环肽。三唑和β-丙氨酸为连接剂的双环肽在人卵巢腺癌细胞中的细胞传递能力提高了7.6倍和19.3倍。而2个母体单环肽[R5]和[WR]4仅能分别提高1.3倍和3.7倍。这种双环肽可以作为一类新的细胞穿透肽和细胞递送工具,在药物递送方面有进一步的应用价值。
传统的双环肽获得方法大多数是使用烷基化试剂对Cys进行修饰,虽然这些方法取得了很大的成功,但这种方法既不是选择性的,也不是生物相容性的。Ullrich等[46]提出了一种肽双环化的新技术,该策略不需要催化剂,具有生物相容性,并正交于所有的典型氨基酸。该策略是基于1,2-氨基硫醇和2,6-二氰吡啶之间的选择性缩合反应,可以直接获得高收率的复杂双环肽。使用此策略,还可以在双环肽的基础上进一步合成三环肽。
然而,随着双环肽序列的增加,分子的刚性往往难以维持,特别是对于一些缺乏二级结构和疏水核心的双环肽,这在很大程度上限制了双环肽与靶标结合的亲和力。Lin等[49]发现了一种胱氨酸桥和脯氨酸残基约束的双环肽新支架,该支架具有良好的刚性,双环肽可以通过该支架稳定成有序结构。
4 双环肽库的构建
随着双环肽市场的增加,单纯的合成方法已经不能满足如此大的需求量,因此从肽库中筛选是一种高效可行的方法,双环肽库的合成由此产生。双环肽库可以由化学法和生物法2种方法构建。
4.1 化学法构建双环肽库
正如前面所提到的,Sun等[33]利用树脂上分子内硫酯连接和树脂外DMSO介导的二硫形成,构建了第1个化学合成的含有9个双环肽的小文库。
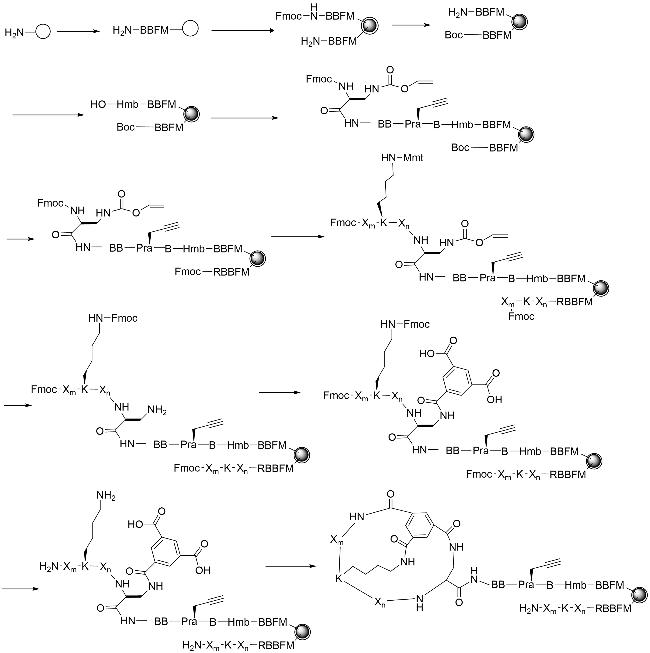
然而,在筛选OBOC文库后,无法确定命中的双环肽的结构[9]。针对这一缺点,Joo等[53]报道了一株双化合物法(OBTC)。在OBTC文库中,树脂珠分离成2个不同的层,珠粒表面显示1个环状肽,用于筛选大分子受体,而内部包含1个相同序列的线性肽,可作为编码标签。在对化合物库进行筛选时,目标靶点只与外层的双环肽结合,内部的线性序列不对其产生干扰,筛选得到的双环肽可以通过Edman降解或质谱方法进行测序。在OBTC文库的构建中,双环肽的肽环化是由刚性小分子支架、均苯三聚酸(TMA)、N端胺、单甲氧基三苯甲基(Mmt)保护赖氨酸和C端L-2,3二氨基丙酸(Dap)侧链之间形成的3个酰胺键介导的[54]。
Lian等[55]在ChemMatrix树脂上合成了OBTC文库,以用来筛选针对蛋白酪氨酸磷酸酶1B(PTP1B)的特异性抑制剂,但所得到的命中肽具有较差的细胞穿透性。随后,研究人员将环肽与环细胞穿透肽融合产生的双环肽具有较好细胞渗透性,并保留识别特定细胞内靶点的能力。该方法在肽基-脯氨酸顺式-反式异构酶(Pin1)细胞渗透性抑制剂的测试中仍然起效,具有一定的普适性。
4.2 生物法构建双环肽库
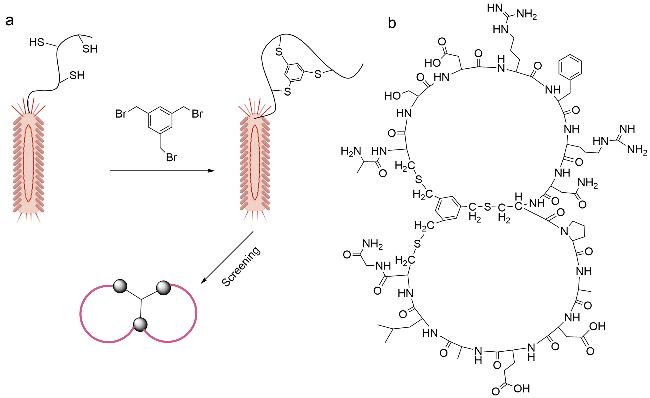
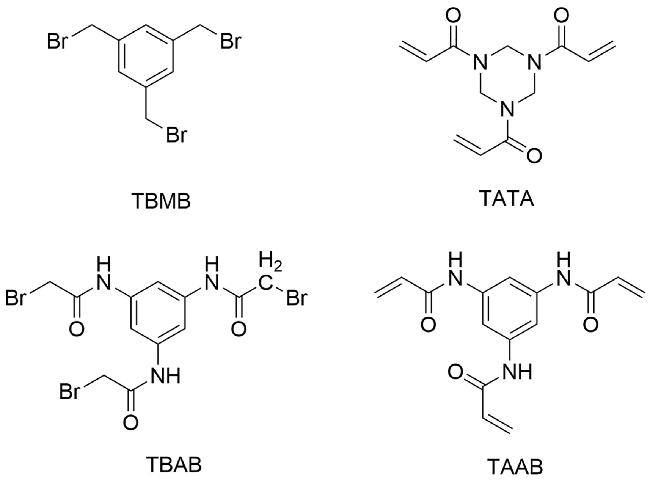
双环肽文库已被证明具有巨大的应用潜力与研究价值。在学术界与工业界,各种双环肽文库已被成功应用于特定目标分子的筛选、药物先导化合物的发现以及分子间的相互作用等研究中。
5 双环肽的应用
由于双环肽具有较好的靶点亲和力、较高的稳定性以及较好的细胞膜通透性,使得双环肽在临床上的应用具有很好的前景,双环肽具有各种各样的生物活性,例如抗肿瘤、抗菌、抗血管生成、抗凝血等。
5.1 双环肽偶联物(靶向给药)
将双环肽与药物、毒素形成偶联物进行靶向给药的方法已经成为肿瘤治疗学中颇具前景的手段。双环肽偶联物的概念源自抗体-药物偶联物(ADCs),ADCs已经成为改善小分子药物药效学的主流手段,ADCs是指一种高效的小分子治疗药物与针对特定靶点的单克隆抗体相偶联,形成抗体-药物复合物[62]。然而,ADCs存在着一些缺点,主要包括:1)抗体具有一定的免疫原性[63],可能会引起一些免疫反应,包括过敏反应、呼吸道症状以及过敏性低血压等[64],因此在使用ADCs时,建议与抗组胺药等配伍使用;2)单克隆抗体的尺寸过大(约150 kDa),使得ADCs难以进入细胞内发挥作用[65],据估计,仅有约0.1%的药物可以到达肿瘤组织,这是ADCs最为致命的缺点;3)ADCs具有较长的体内半衰期[66],较长的暴露时间可以弥补其较差的组织穿透性,但长时间暴露可使它们易被循环血浆蛋白酶酶解,逐渐地全身负荷释放[67],从而易引起全身性副作用;4)抗体通常通过肝脏代谢消除,导致有效载荷在肝脏和胃肠道中释放,因此肝脏和胃肠道毒性是ADCs常见的毒副作用[68]。双环肽偶联物的出现很好地解决了ADCs上述缺点。首先,双环肽具有2个肽环,都可以参与结合,具有更高的亲和力。同时分子质量比抗体小很多,组织穿透能力大大提高,能够快速、有效地将药物递送到肿瘤组织中。其次,双环肽配体易于合成和偶联,在开发过程中更加省时省力。最后,肽的特性使体内半衰期缩短且大部分经肾脏消除,限制了人体对有效载荷的暴露,最大限度地减少所偶联的药物、毒素对正常组织的损害[69]。同时由于双环肽降解产物是氨基酸,几乎对人体不会产生毒性影响。基于这些优点,我们认为双环肽在偶联靶向治疗领域的应用前景十分广阔。目前双环肽偶联物已经成为Bicycle Therapeutics公司的平台技术,用于选择性递送化疗药物或其他药物以靶向病灶。双环肽偶联物可以进一步细化为双环肽药物偶联物(BDCs)、双环肽毒素偶联物(BTCs)和肿瘤靶向免疫细胞激动剂(TICAs),三者在本质上并无差别,仅是偶联物不同。
5.1.1 双环肽药物偶联(BDCs)
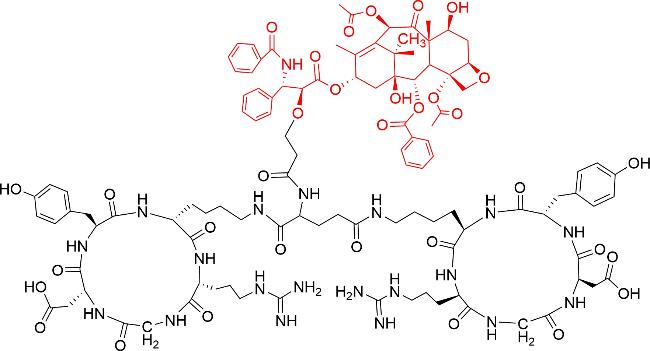
5.1.2 双环肽毒素偶联(BTCs)
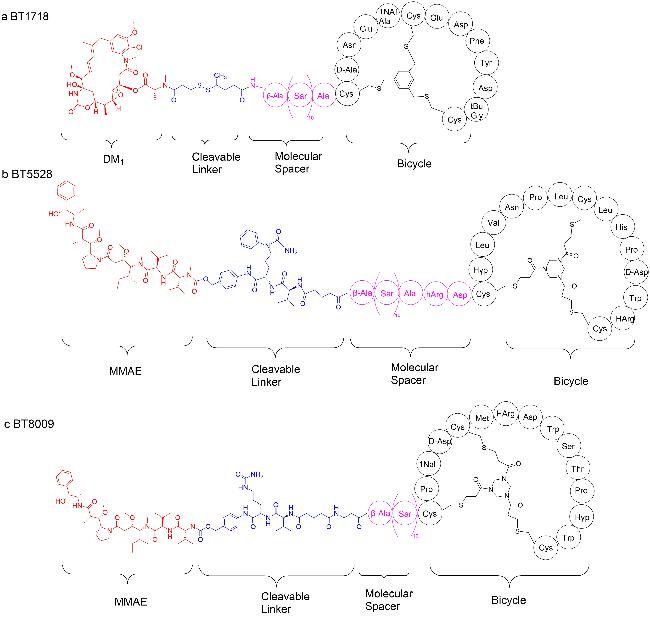
表1 BTCs总结Table 1 Summary of BTCs |
| Name | Structure | Application | Target | Coupling | Linker | Clinical trial phase |
|---|---|---|---|---|---|---|
| BT1718[71-73] | Scheme 9 a | Non-small cell lung cancer、Breast cancer and other solid malignancies | MT1-MMP | DM1 | Disulfide bond | Clinical Phase II |
| BT5528[74-75] | Scheme 9 b | Ovarian and uroepithelial cancers | EphA2 | MMAE | Val-Cit | Clinical Phase II |
| BT8009[69,76] | Scheme 9 c | Advanced solid tumours associated with Nectin-4 expression | Nectin-4 | MMAE | Val-Cit | Clinical Phase I |
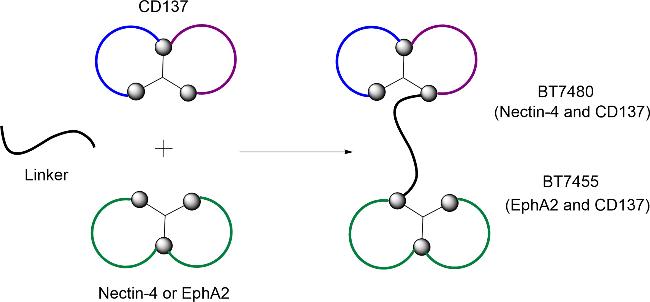
5.1.3 肿瘤靶向免疫细胞激动剂(TICAs)
CD137是一种免疫共刺激受体,属于肿瘤坏死因子(TNF)受体超家族的一员,主要参与T细胞、自然杀伤细胞和其他免疫细胞的激活[77],CD137可延长T细胞抗肿瘤活性、增殖活性、存活周期等。肿瘤靶向免疫细胞激动剂(TICAs)主要有2种——BT7480和BT7455,两者均是CD137激动剂。BT7480[78](图2)是一种高效的Nectin-4依赖性CD137激动剂,于2021年进入临床试验。BT7480临床试验数据上的成功,激励了研究人员对TICAs的进一步开发,随后,开发了第2种TICAs——BT7455。BT7455[79](图2)是一种高效的EphA2表达依赖性CD137激动剂,在临床前研究中对EphA2阳性癌症提供高效的CD137激动作用,具有最佳的靶点结合、药理学和药代动力学特性,将进入I期临床的研究中。
5.2 PPIs
PPIs在众多生理过程中发挥着重要作用,选择性调节PPIs已成为一种新的治疗干预策略。由于PPIs靶点具有宽大、平坦而无特征的结合表面,小分子化合物很难与其结合。肽类药物在针对PPIs方面更具优势,双环肽相比线性肽和单环肽,对PPIs的亲和力和特异性更高,因此引起了研究人员极大的兴趣。
表皮生长因子(EGF)已被证明参与许多类型的实体肿瘤,包括头颈部癌、乳腺癌、结肠癌、卵巢癌和非小细胞肺癌[80]。因此,EGF-EGFR通路已经成为治疗肿瘤的焦点,尽管临床上已经存在EGFR抑制剂,但大多数患者会出现耐药性,导致长期用药疗效的降低。并且,靶向EGFR的药物由于生物利用度和毒副作用的原因,使用非常受限。在这种情况下,Guardiola等[81]研究发现,直接抑制EGF可能会产生更好的治疗效果,而多肽类药物可以实现此目标。随后该课题组提出了基于结构的双环约束肽的设计,并模拟了EGFR的一个相互作用域[82]。除了在分子水平上研究它们与EGF的相互作用外,该课题组还在一个特定的受体配体实验中证实了它们阻断EGF-EGFR相互作用的潜力,并且在过表达EGFR的人类癌细胞中也被证实了治疗潜力。
从肽库中筛选PPIs是一种快捷高效的方法,Lian等[83]首先选择平面结构作为支架,合成了双环肽库。平面支架使得到的双环肽偏向于整体的平面几何形状,使分子的表面积最大化,从而能够与平坦的蛋白质表面相互作用,这种类型的双环肽可能为抑制PPIs提供了一种通用的解决方案。接着该课题组对肿瘤坏死因子α(TNFα)双环肽文库的筛选发现了抑制TNFα-TNFα受体相互作用的有效拮抗剂Anticachexin C1,并保护细胞免受TNFα诱导死亡。
抑制p53与鼠双微体2(murine double minute MDM2)和鼠双微体X(murine double minute X MDMX)相互作用的策略被证明是一种很有前景的肿瘤治疗方法[84]。而利用双环肽抑制p53和MDM2/MDMX之间的PPIs具有更大的优势,Li等[85]采用全碳氢化合物钉接策略与内酰胺相结合的方法合成了双环钉肽p53-16(图式10),显著改善了α-螺旋度和蛋白水解稳定性。P53-16对MDM2和MDMX具有纳米级的结合亲和力,可以穿透细胞膜,激活p53通路选择性抑制肿瘤细胞的活性。RAS是抗肿瘤药物的一个有吸引力的靶点,但是,作为参与细胞内PPIs的蛋白,RAS一直是一个非常具有挑战性的靶标。Trinh等[86]筛选针对G12V突变体KRAS的双环肽库并进行命中优化,产生了一种中等效价的细胞渗透性KRAS抑制剂,可以物理阻断RAS效应物的相互作用,诱导癌细胞的凋亡死亡。可以此为先导化合物,进行进一步的结构优化,为研究RAS抑制剂提供了有力的工具。
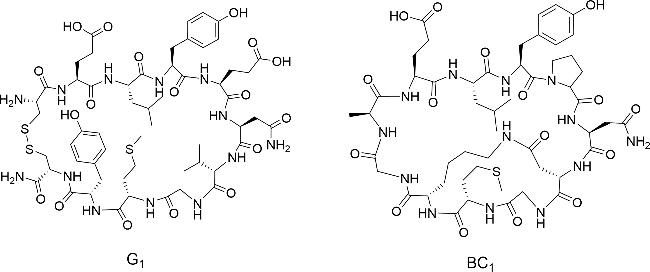
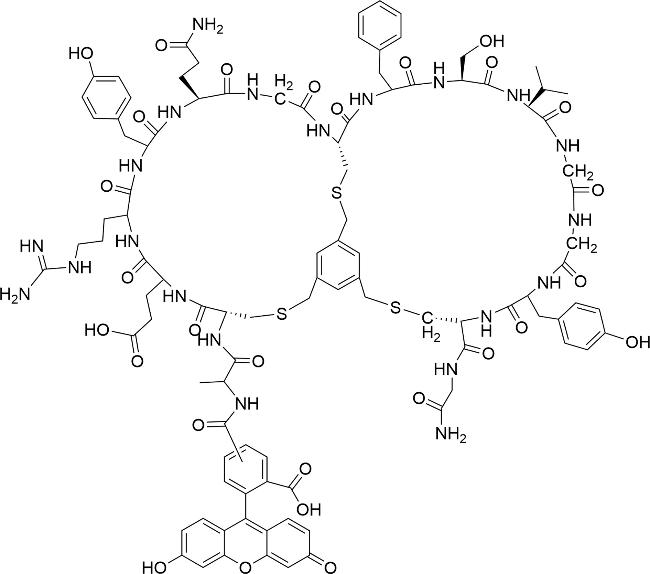
肽G1(图式11)是由11个氨基酸残基组成的肽大环,靶向生长因子结合蛋白2(Grb2)的Src同源性 2(SH2)结构域。Quartararo等[87]以G1为起点,通过钉接肽的方法来生产具有更高亲和力、选择性和抗降解性的双环肽。经过2轮迭代设计产生双环肽BC1(图式11),其效力是G1的60倍,选择性是G1的200倍。而且,在G1完全降解的条件下,在缓冲的人血清中,BC1在24 h后是完全完整的,48 h后降解率低于15%。这说明BC1的抗降解能力非常强,可在体内达到长效的效果。这种肽循环方法有望开发出SH2结构域和其他磷酸酪氨酸(pTyr)结合蛋白的选择性抑制剂,以及许多其他PPIs抑制剂。
虽然有许多双环肽对PPIs显示出来很好的亲和力和活性,可作为先导化合物进行进一步的开发,但对细胞膜的渗透性一直是一个棘手的问题。目前,常用的解决方法是双环肽与CPPs偶联或结合,环状CPPs代谢稳定,可大大提高了胞质进入效率。
5.3 酶抑制剂/激动剂
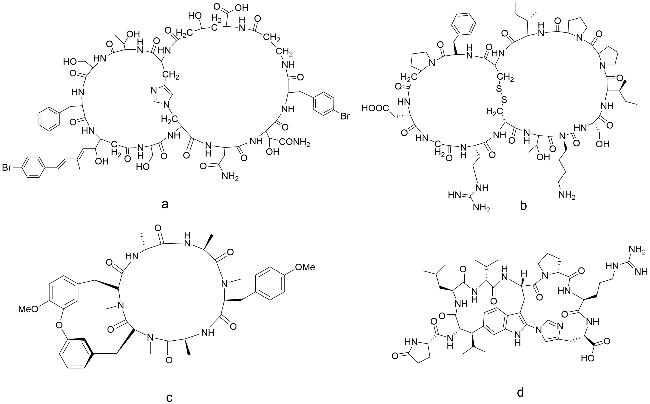
然后,主要讨论酶抑制剂。(PKal)是激肽-激肽系统的一员,是一种丝氨酸蛋白酶,它能催化生物活性肽-缓激肽的释放,从而引起炎症、血管舒张、血管通透性增加和疼痛。PKal已被确定为治疗糖尿病黄斑水肿(DME)的潜在靶点[89]。采用基于噬菌体展示与化学环化相结合的技术,Teufel等[90]已经确定了具有纳米和皮摩尔效力的高选择性双环肽抑制剂(基本结构见图式13a),通过引入非天然氨基酸和非肽键来增加在生物基质中的稳定性。该多肽在体外抑制缓激肽释放,并在大鼠足部水肿模型和糖尿病诱导的视网膜通透性啮齿动物模型中证实了体内效果,是治疗糖尿病视网膜病变和糖尿病黄斑水肿的有希望的新药物。近期,从双环肽PKal抑制剂类似物文库中筛选了一种有效且稳定的抑制剂。这种药物被命名为THR-149,临床前实验表明,该双环肽在糖尿病大鼠模型中被证明可以预防糖尿病引起的视网膜渗漏。Van等[91]通过一系列临床前实验进一步表明,反复给药THR-149可以减少糖尿病大鼠的几种与DME相关的关键病理,如视网膜增厚和神经细胞破坏。这一实验证实了THR-149可进一步进行临床开发,有望成为治疗糖尿病的新药物。
尿激酶型纤溶酶原激活物(uPA)是一种胰蛋白酶样丝氨酸蛋白酶,参与细胞外基质(ECM)蛋白的周转,与肿瘤生长和侵袭有关,目前很难研发出针对uPA的高效特异性小分子抑制剂,而大分子抑制剂,例如单克隆抗体,穿透肿瘤细胞的能力会很差。因此,肽类药物可以作为重点开发对象。Angelini等[92]从肽组合库中分离出双环肽抑制剂——UK18(图式13b),对人类uPA的结合亲和力比之前分离得到的最佳单环肽upain1高200倍。UK18作为小的高约束双环肽(<2 kDa),既有典型的蛋白质特性(与靶标具有较大的相互作用界面),又有良好的结合亲和力和特异性。Harman等[93]通过筛选高度多样化的噬菌体展示文库,得到了一系列限制性双环肽,用于进一步设计具有更高亲和力和更高抑制血管紧张素转换酶(ACE2)活性的双环肽。这种新型的双环肽ACE2抑制剂是迄今为止在体外最有效的ACE2抑制剂之一,将成为进一步探索ACE2功能和潜在疗效的有力工具。凝血因子XII(FXII)抑制剂对研究内在凝血途径中的蛋白酶、抑制凝血试验中的接触活化具有重要意义,并且在抗血栓治疗中具有潜在的应用前景。然而,迄今为止开发的合成FXII抑制剂具有较弱的结合亲和力和/或较差的选择性。Baeriswyl等[94]生成并筛选了包含数十亿个结构多样的双环肽的新型组合文库,以鉴定合成的FXIIa抑制剂。最佳双环结构FXII618(图式13c)抑制常数Ki为22 nmol/L,选择性超过其他蛋白酶2000倍,有效地和选择性地抑制了血浆和全血内在凝血途径的启动,而不影响外在途径。蛋白酪氨酸磷酸酶(PTPs)介导许多细胞过程的执行和调节,例如信号转导,而PTPs抑制剂的设计一直以来都是具有挑战性的目标,因为所有PTPs都具有高度保守的活性位点结构,该结构带正电,需要带负电的部分才能紧密结合。但是,带负电的物质通常不被细胞膜渗透。Liao等[95]直接对组合文库进行筛选,开发了针对T细胞PTP(TCPTP)的细胞渗透性双环肽基抑制剂PTP1B(图式13d),其一个环具有细胞穿透基序(CPP),第2个环具有靶标结合序列,该双环肽有望开发为PTPs抑制剂。
5.4 受体抑制剂
Notch信号的失调与包括癌症在内的几种人类疾病有关[97]。因此,靶向Notch的药物开发具有很大的前景,Urech等[98]利用噬菌体展示技术从组合文库中分离出对人Notch1受体NRR具有高度亲和结合力的双环肽FL-NRR17(图式14)。表皮生长因子受体Her2的异常表达与包括乳腺癌在内的多种恶性肿瘤有关。Diderich等[99]通过对66个双环肽库进行筛选,得到了若干针对表皮生长因子受体Her2的双环肽特异性配体。获得的最佳双环肽与Her2结合,KD(平衡解离常数)值为304 nmol/L。不幸的是,在细胞实验中,上述双环肽配体并没有表现出希望的活性,但这些肽配体可用于调节其他疾病靶点的构象,具有潜在应用于肿瘤成像和治疗的价值。
5.5 抗菌双环肽
2019年,Imai等[103]通过筛选Photorhabdus分离物得到了一种新的抗生素——Darobactin,具有独特的双环结构。该化合物通过靶向外膜蛋白BamA对多种革兰氏阴性病原体非常有效,且对常见的共生肠道细菌和人类细胞系没有活性,这就意味着对人体影响小,安全性高。Adaligil等[104]使用“镜像噬菌体展示技术”[105]发现了由D-氨基酸组成的肽抗生素,其中,活性最强的双环结构P14(图式15a)对金黄色葡萄球菌和MRSA具有良好的抗菌作用,MIC值分别为8 μg/mL和32 μg/mL。虽然一些天然或设计的抗菌肽对哺乳动物细胞具有细胞毒性,但双环抗菌肽P14在质量浓度高达256 μg/mL时,既不会引起红细胞的溶解,也不会对哺乳动物细胞显示出明显的毒性。这表明P14可作为一种有潜力的抗菌双环肽被进一步开发。
虽然抗菌肽较传统抗生素的耐药性有所提高,但细菌产生抗药性是不可避免的,不断研发新的抗菌化合物和发现新的抗菌作用机制将有助于限制抗生素耐药性。同时,严格限制在非必要病例中使用抗菌药物,合理与不同类型的抗菌药物连用,将进一步限制耐药菌的风险。
5.6 成像和造影
在医学诊断学研究中,分子成像技术处于非常重要的地位。多肽分子经标记后成为一类分子探针,可用于疾病诊断、治疗和愈后的追踪。双环肽是非常有吸引力的正电子发射计算机断层显像(PET)示踪剂和其他诊断显像剂。双环肽具有快速肿瘤穿透特性,加上高效和化学多样性,可以产生出色的信号与背景比、更短的给药和成像之间的延迟。因此为临床诊断提供了高对比度成像探针,在靶向治疗中具有令人信服的额外潜力。
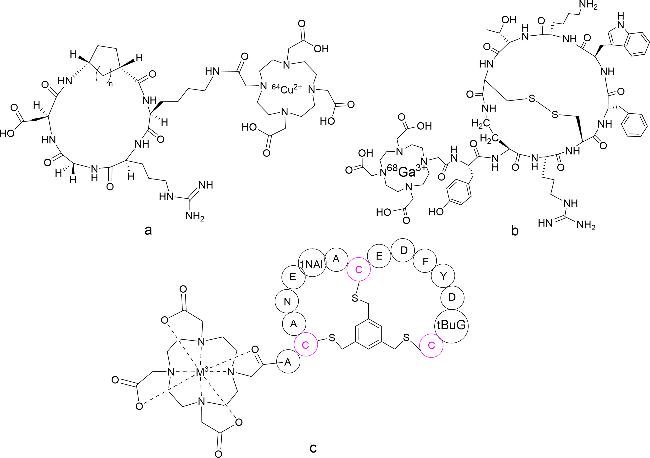
为了检测早期肿瘤细胞,必须将肿瘤靶向肽(如RGDs)结合到成像探针中。Park等[110]在四肽(RGDK)序列上接枝氨基环戊烷(ACP)和氨基环己烷(ACH),合成了2个新的双环RGD肽,并与DOTA偶联(图式16a)。采用放射性金属64Cu标记这些偶联物,2种偶联物对U87MG胶质母细胞瘤细胞均表现出较高的亲和性,且亲和性优于单环c(RGDyK),可作为PET的显像剂。Fani等[111]设计了一种双环生长抑素类似物,采用177Lu和68Ga标记的DOTA(1,4,7,10-四氮杂环十二烷-1,4,7,10-四乙酸)偶联双环类似物,对表达生长抑素受体亚型2和3的肿瘤异种移植物进行体外内化、外排、药代动力学和影像学研究。在相应的裸鼠模型上进行生物分布和PET/CT研究,最佳偶联物68Ga-AM3(图式16b)注射后1 h的PET/CT表现为肿瘤定位清晰,肾脏可见,背景可忽略,可成为一种极好的成像示踪剂,尤其是PET示踪剂。Melemenidis等[112]合成了一个对αvβ3具有高亲和力的环状RGD变体[c(RGDyK)],并将其多价偶联到氧化铁微粒(MPIO)上。在体外流动条件下,测试了c(RGDyK)-MPIO与表达αvβ3的内皮细胞的结合特性,随后在结肠癌和黑色素瘤动物模型中量化了c(RGDyK)-MPIO在体内的造影效果,可以为早期肿瘤检测和确定特定治疗的适用性提供机会。Eder等[113]以肿瘤过表达的基质金属蛋白酶MT1-MMP为靶标,鉴定出一种对MT1-MMP具有亚纳摩尔亲和力的双环肽,通过N端与螯合剂DOTA偶联,进行多次实验后,得到最佳显像剂BCY-C2(图式16c),体内研究显示小鼠模型的肿瘤摄取高。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
对于可成像的双环肽偶联物,可通过提高肽的蛋白水解稳定性以及通过脂肪酸偶联延长血清半衰期后,肿瘤摄取进一步增加。当然,通过改变分子间隔物的位置、组成和大小,以及通过使用与白蛋白具有一定亲和力的脂肪酸衍生物,可以进一步优化肿瘤信号和体内选择性。这些例子都有力地说明,双环肽在成像和造影方面有很大的应用价值。
6 展望与讨论
双环肽领域快速发展,越来越得到人们的关注。本文从双环肽合成和应用2个方面进行了讨论,对双环肽的现状和未来的发展进行系统地总结与展望。
在双环肽的获取方面,各种合成方法和肽库筛选相结合,获取途径进一步扩大,简便、易得是双环肽的一大优点。含有二硫键的肽在自然界中广泛存在,二硫键赋予了双环肽良好的热稳定性和水解酶稳定性,因此,二硫键介导的双环肽合成是目前常用的策略。基于二硫键的不稳定性,现多采用其他小分子连接物为介导,最常见的有硫醚键、烯烃、炔烃等。小分子介导的双环肽是化学和生物合成双环肽常用的手段,开发结构更稳定、生物相容性更高、毒性更低的小分子连接物是目前双环肽合成领域的热点。除了小分子连接物的介导以外,分子内酰胺键、头尾相连等简便方法也被广泛应用。然而,双环肽合成仍然面临一些挑战,例如,合成效率低、操作烦琐和大规模合成成本高昂等。在未来,开发效率更高、成本更低的合成方法是重要的研究方向。
在双环肽的活性方面,首先,双环肽具有更高的亲和力、稳定性和细胞膜通透性。因此,未来可以将具有一定活性的线性肽、单环肽进行双环化以提高活性和生物利用度;第二,相对于抗体的分子量大、免疫原性等缺点,ADCs的缺点能很好地被双环肽偶联物解决,我们期待双环肽偶联物的成功上市,这一领域也将是未来双环肽靶向治疗的热点。基于双环肽所表现出来的一系列优点,我们大胆推测,在将来,也许双环肽会取代抗体作为治疗、诊断试剂方面的应用。第三,对于小分子药物,一些被认为“不可成药的靶点”,双环肽是可以实现的,这有望靶向一些棘手的靶点,对治疗一些人类尚未解决的疾病存在潜在价值。然而,双环肽仍然存在一些需要进一步克服的缺点。首先,肽类药物的溶解度一直是一个重要的问题,溶解度直接关乎药物的细胞渗透性和生物利用度。虽然目前可以采用制剂的方法解决部分溶解性问题,但想要完全解决这一问题又不影响活性,还需要研究人员进一步的探索。其次,双环肽较线性肽和单环肽的构象刚性增加,与受体结合时熵罚降低,有利于结合靶标。但更大的刚性意味着更低的自由度,这就有可能导致分子与靶点结合时的构象可能处于不利的能量和空间位置,从而导致脱靶现象。最后,虽然部分双环肽对受体具有很高的亲和力,但在活性实验中并没有表示出预期的活性,这是未来双环肽需要攻克的重点。
虽然双环肽仍然具有一些缺点,但这并不能否定双环肽在药物开发中的巨大潜力。我们相信,未来,双环肽很有可能会成为医药发展的强大驱动力,受到越来越多的关注。