



1 引言
2 氧气电还原反应基本原理
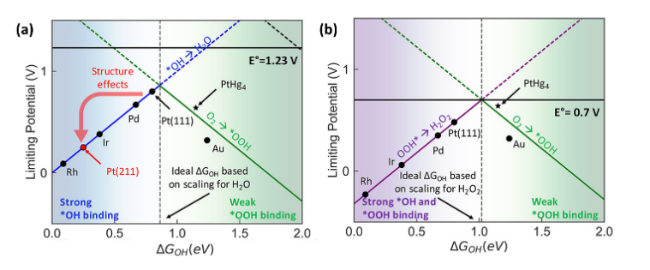
图1 经典ORR结合能-理论极限电位火山图[14]。(a)在四电子反应路径中不同金属*OH结合能与ORR理论极限电位的关系,其中蓝色实线表示强结合*OH区域,绿色实线表示弱结合*OOH区域;(b)在二电子反应路径中不同金属*OH结合能与ORR理论极限电位的关系,其中紫色实线表示强结合*OOH区域,绿色实线表示弱结合*OOH区域Fig. 1 The classical volcano plot with binding energy and theoretical limiting potential[14]. Copyright 2018 American Chemical Society. (a) The relationship between the binding energy of different metal *OH and the theoretical limit potential of ORR in the four electron reaction pathway, where the blue solid line represents the strong binding *OH region and the green solid line represents the weak binding *OOH region; (b) The relationship between the binding energy of different metal *OH and the theoretical limit potential of ORR in the two electron reaction pathway, where the purple solid line represents the strong binding *OOH region and the green solid line represents the weak binding *OOH region |
3 电催化剂的电子结构调控
3.1 化学掺杂工程
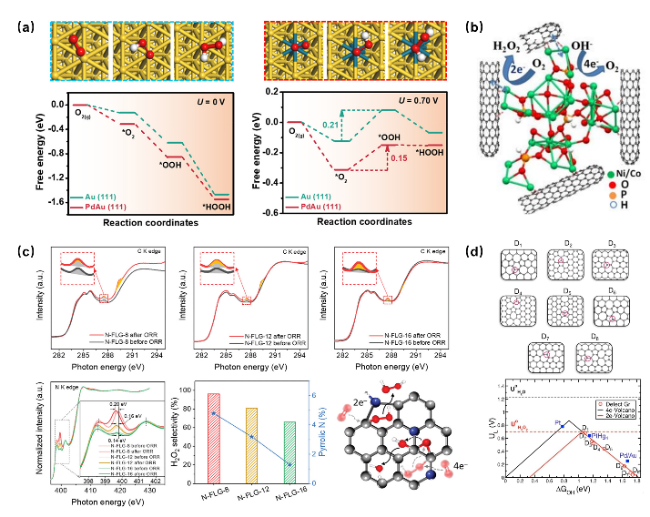
图2 电催化剂的电子结构调控策略促进2eORR合成H2O2。(a)Au(111)和PdAu(111)上的2eORR途径的DFT模拟[17];(b)碳纳米管(CNTs)修饰的亚磷酸镍钴(NiCo−Phi)构型[18];(c)氮掺杂碳基材料反应前后的碳K边和氮K边XANES光谱及不同氮掺杂构型、含量对于ORR反应路径的影响[23];(d)不同碳基缺陷构型对于2电子及4电子ORR火山图的影响[24]Fig. 2 The electronic structure control strategy of electrocatalysts promotes the H2O2 synthesis. (a) DFT simulation of the 2eORR pathway on Au(111) and PdAu(111) [17]. Copyright 2021 American Chemical Society. (b) Carbon nanotubes (CNTs) modified nickel cobalt phosphite (NiCo Phi) configuration[18]. Copyright 2020 Wiley VCH GmbH. (c) The carbon K edge and nitrogen K edge XANES spectra of nitrogen doped carbon based materials before and after reaction, as well as the influence of different nitrogen doping configurations and contents on the ORR reaction pathway[23]. Copyright 2020 Wiley VCH GmbH. (d) The influence of different carbon-based defects on the 2-electron and 4-electron ORR volcano diagrams. Adapted with permission from [24]. Copyright 2018 American Chemical Society |
3.2 缺陷构建工程
4 电催化剂的几何结构调控
4.1 尺寸调控
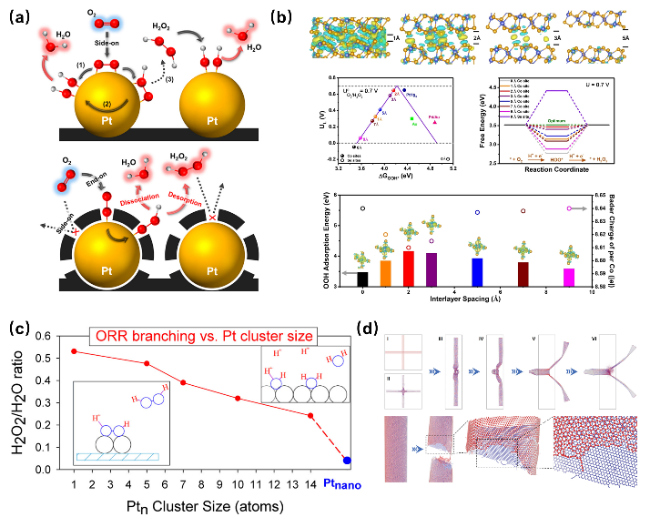
图3 电催化剂的几何结构调控策略促进2eORR合成H2O2。(a)碳涂层调控了O2在Pt表面的吸附模式,其中末端吸附更有利于H2O2的生成[44];(b)CoSe2中不同层间距离对2eORR合成的反应能垒、理论极限电位及中间体吸附能的影响[45];(c)不同Ptn团簇尺寸对于ORR合成的H2O2/H2O比例的影响[39];(d)碳纳米管尖端结构活性位点的形成机制[46]Fig. 3 The geometric structure control strategy of electrocatalysts promotes the H2O2 synthesis. (a) The carbon coating modified on the surface of Pt allows O2 to undergo end configuration adsorption on the Pt surface, thereby facilitating the generation of H2O2[44]. Copyright 2014 American Chemical Society. (b) The influence of different interlayer distances in CoSe2 on the reaction energy barrier, theoretical limit potential, and intermediate adsorption energy of 2eORR synthesis[45]. Copyright 2021 Wiley VCH GmbH. (c) The effect of different Ptn cluster sizes on the H2O2/H2O ratio in ORR synthesis[39]. Copyright 2015 American Chemical Society. (d) The formation mechanism of active sites in the tip structure of carbon nanotubes[46]. Copyright 2023 Wiley VCH GmbH |
4.2 孔隙/层间结构调控
4.3 表面形貌调控
5 电催化剂表面修饰功能化
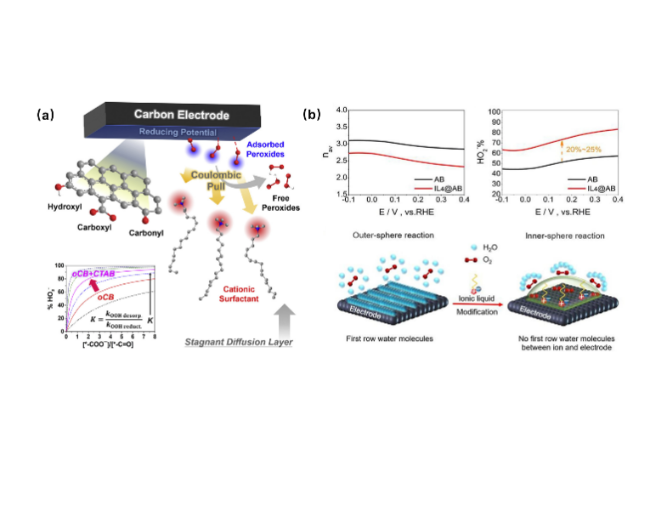
图4 电催化剂的表面修饰功能化策略促进2eORR合成H2O2。(a)在碳基催化剂表面修饰CTAB表面活性剂能够促进H2O2的解吸过程[52];(b)利用离子液体修饰乙炔黑以提高H2O2选择性,其中离子液体中烷基链的延长能够影响碳电极的疏水性[55]Fig. 4 The geometric structure control strategy of electrocatalysts promotes the H2O2 synthesis. (a) Surface modification of CTAB surfactants on carbon-based catalysts can promote the desorption process of H2O2[52]. Copyright 2020 Elsevier Inc. (b) Acetylene black-modified with ionic liquids could improve H2O2 selectivity, and the extension of alkyl chains in ionic liquids can affect the hydrophobicity of carbon electrodes[55]. Copyright 2020 Elsevier B.V |
6 原子级活性位点设计
6.1 金属活性中心调控
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
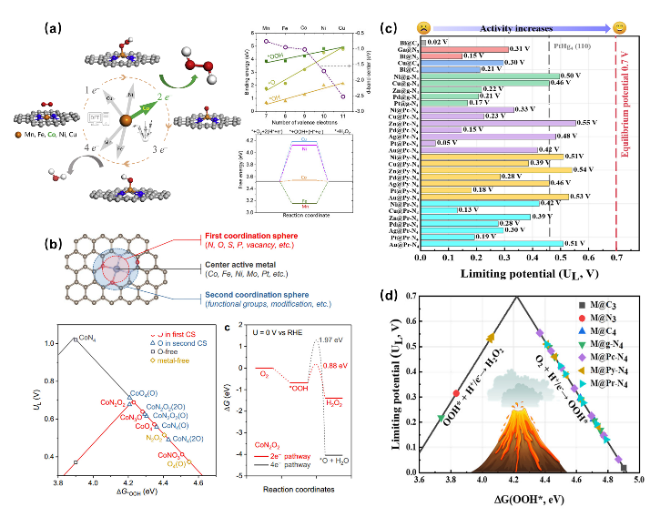
图5 电催化剂的原子级活性位点策略促进2eORR合成H2O2。(a)不同金属活性中心对于M—N—C型单原子催化剂d带中心及自由能垒的影响[58];(b)Co—N—C单原子催化剂中的第一配位域及第二配位域,其中掺杂氧原子及修饰C—O—C含氧官能团有利于2eORR反应能垒的降低[67];不同金属活性中心对于M@PC-N4型超分子催化剂(c)2eORR火山图及(d)理论极限电位的影响[64]Fig. 5 The geometric structure control strategy of electrocatalysts promotes the H2O2 synthesis. (a) The influence of different metal active centers on the d-band center and free energy barrier of M—N—C type single atom catalysts[58]. Copyright 2020 Elsevier Inc. (b) The first and second coordination domains in Co—N—C single atom catalysts, where doping oxygen atoms and modifying C—O—C oxygen-containing functional groups are beneficial for reducing the energy barrier of the 2eORR reaction[67]. Copyright 2021 American Chemical Society. The influence of different metal active centers on the (c) 2eORR volcano plot and (d) theoretical limiting potential of M@PC-N4 supramolecular catalyst[64]. Copyright 2019 American Chemical Society |