



1 引言
甘油常规的应用领域包括化妆品、个人护理产品、制药、溶剂、食品添加剂和烟草等[3]。另外,源于生物柴油的蓬勃发展,甘油已经成为最重要的生物质平台化合物之一,通过不同转化途径(如 生物发酵、氧化、氢解、脱水、氯化、酯化、醚化、羰基化、聚合等)生产增值化学品和中间体(如甘油醛、二羟基丙酮、甘油酸、乳酸、丙烯酸、丙二醇等)[4,5]。目前,甘油氢解和选择性氧化是最为活跃的领域。例如,甘油氢解制备聚酯树脂化学原料1,2-丙二醇已经实现产业化,许多学者正致力于甘油氢解或发酵法制1,3-丙二醇(聚对苯二甲酸三 甲酯单体)的工业应用。而在甘油选择性氧化方面,甘油氧化生成二羟基丙酮、羟基丙醛、甘油酸和甘油醛的工艺也日趋成熟。近年来,随着可降解塑料的推广,聚乳酸材料备受关注,这使得甘油催化氧化制备乳酸变成极具吸引力的转化路线。目前,乳酸主要通过生物基碳水化合物(如葡萄糖和蔗糖)经生物发酵产生[6]。该方法需要不断添加碱性介质以保持反应环境适宜的pH,避免酶催化剂的失活。同时,反应完成后需要H2SO4对乳酸盐酸化,导致形成大量的盐废物,生产成本高、环境污染问题严重[7,8]。以甘油为原料制备乳酸,被认为是微生物发酵最具潜力的替代方案[1,9]。
2 甘油转化制乳酸的反应机理
2.1 甘油水热转化制乳酸
Shen等[18]进一步证明了碱金属氢氧化物(KOH、NaOH、LiOH)和碱土金属氢氧化物 (Ba(OH)2、Sr(OH)2、Ca(OH)2、Mg(OH)2)以及两性氢氧化物Al(OH)3催化甘油水热转化制乳酸的可行性。除Al(OH)3外,上述碱性催化剂均能催化甘油生成乳酸或乳酸盐,碱金属氢氧化物的效果优于碱土金属氢氧化物。KOH在较低的浓度或较短的反应时间内的效果优于NaOH,说明碱金属离子 种类影响反应行为。Sharninghausen等[19]对比KOH、LiOH和NaOH的性能,发现KOH是生成乳酸的最佳催化剂。Chen等[21]将CaO作为固体碱用于甘油转化制乳酸,反应路径如图1所示。甘油首先氧化为甘油醛,之后甘油醛脱水生成2-羟基丙烯醛,后者很 容易通过酮-烯醇同分异构作用转化为丙酮醛。同时,2-羟基丙烯醛和丙酮醛可能与中间产物H2发生加氢反应,生成少量的丙二醇。最后,在CaO催化作用下,丙酮醛容易发生二苯乙醇酸重排反应形成乳酸钙(图1A)。另外,氧化物离子形成过程中也可能演变成寡聚(图1B)。这些低聚物可溶性较差或被吸附在催化剂表面,导致反应效率降低。因此,单独使用固体碱催化剂反应活性较低,进一步应用受到限制。
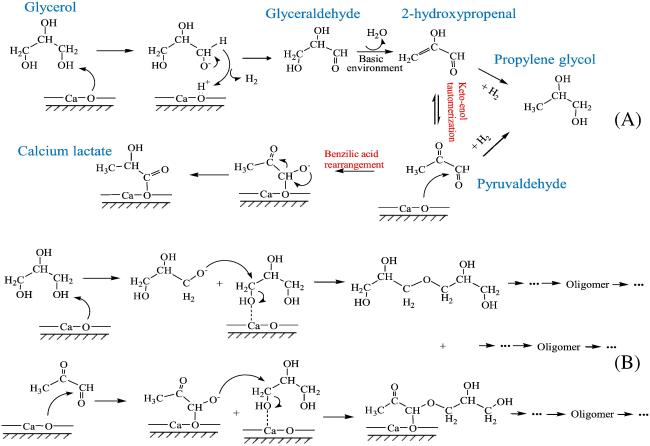
水热法反应体系反应条件苛刻、废弃物排放量大,且产品和催化剂分离成本高、工业应用难以推进。目前,甘油转化制乳酸主要聚焦在多相催化体系的开发。
2.2 甘油选择性氧化制乳酸
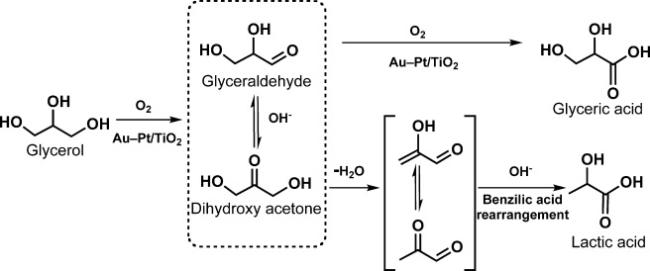
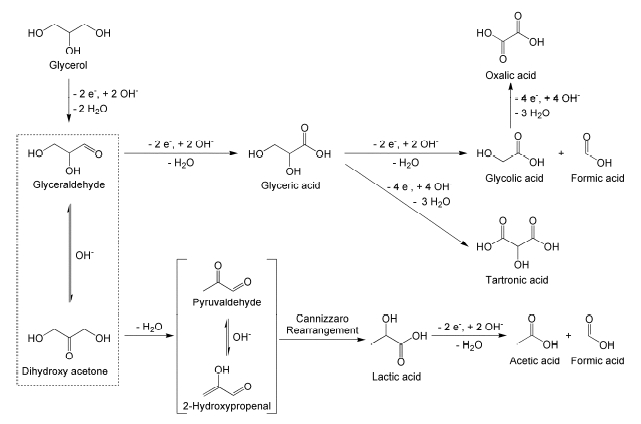
除了甘油水热转化,引入氧化剂和催化剂是另一条甘油制乳酸的可行路径。目前,多数研究工作者主要聚焦研究温和条件下贵金属(Au、Pt、Pd)催化剂催化甘油选择性氧化反应[22,23]。但该体系通常需要添加一定量的碱(NaOH、LiOH或KOH)来提高反应体系的pH,耦合碱在水热条件下的催化脱氢和脱水能力,提高催化活性和选择性。2010年,Shen等[15]报道了以Au-Pt/TiO2为催化剂一锅法将甘油高效转化为乳酸,反应路径如图2所示。即甘油首先在O2存在下氧化脱氢生成中间体二羟基丙酮(Dihydroxyacetone,DHA)或甘油醛(Glyceraldehyde,GLA),DHA和GLA可以相互转化,之后DHA再经过催化脱水生成丙酮醛(Pyruvialdehyde,PA),最后PA经二苯乙醇酸重排生成LA。DHA和GLA之间的平衡取决于pH,不添加碱时氧化产物为GLA、DHA和甘油酸(Glyceric acid,GLYA),说明碱存在促进Au-Pt催化甘油氧化制乳酸,这与Kishida等[17]报道的结果一致。碱的存在一方面促进甘油羟基的去质子化,另一方面加速中间产物(DHA)向乳酸的转化。
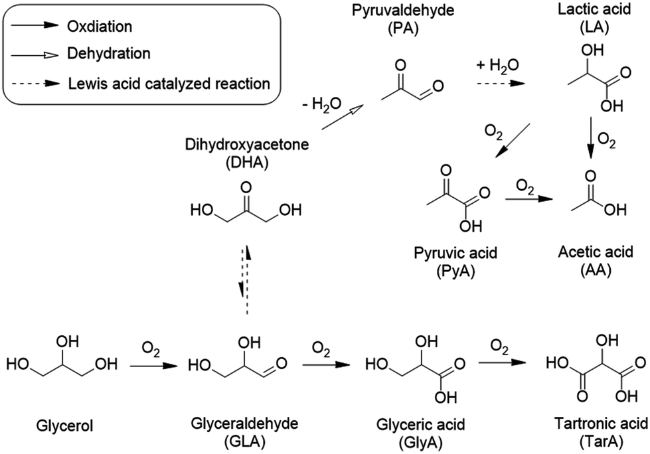
如前所述,碱溶液会导致乳酸盐的形成,生产乳酸需要额外的反应和分离步骤,不仅增加了生产成本,而且产生了大量的盐废物。因此,无碱体系下,甘油选择性氧化制乳酸更具有应用潜力。研究表明,与碱类似,Lewis酸也能够在甘油转化中起到催化脱水和异构化的作用。Xu等[24]同样以AuPt/TiO2为催化剂,在Lewis酸AlCl3存在下,发现甘油可以在无碱条件下选择性地将甘油直接氧化为乳酸。生成乳酸的反应路径为:甘油氧化脱氢生成DHA和甘油醛中间体,H+和Al3+协同催化这些中间体脱水并重排为LA。但是,Al3+的水化作 用和Cl-的毒性往往影响它们的实际应用。2014年,Cho等[25]提出了一种双功能催化剂的策略,采用Pt/Sn-MFI催化剂,实现甘油直接氧化制乳酸(图3)。在温和的反应条件下,甘油首先在Pt金属活性位上被氧化生成DHA或GLA,之后在Sn-MFI分子筛Lewis酸的作用下,GLA快速转化为较为稳定的DHA,DHA再经脱水生成PA,PA与水加成后发生分子内氢转移生成LA。Tao等[26]提出将Lewis酸位点Al3+或Cr3+通过金属离子交换质子引入到多金属氧酸盐(POMs)中,制备具有Lewis酸活性位和氧化活性位的双功能催化剂(AlPMo或CrPMo),直接催化甘油氧化制乳酸。研究发现,Lewis酸金属离子对POMs的氧化还原电位有显著影响,Lewis酸性越强,其氧化还原电位越高,从而提高了甘油转化率。同时,较强的Lewis酸催化剂提高了对DHA的化学选择性,而DHA脱水以及PA到LA的转化过程同样也需要Lewis酸位点作用。除此之外,AlPMo或CrPMo催化效率较高也与其自身疏水性有关,不溶性的AlPMo或CrPMo能够从二 级结构中快速释放生成的水,促进DHA脱水为PA,呈现出LA快速生成的反应特点。Feng等[27]以层 状L-Nb2O5为载体制备了Pt/L-Nb2O5双功能催化剂,同样实现了甘油直接转化制乳酸。甘油首先通过Pt纳米颗粒催化转化为DHA和GLA,DHA在Brønsted酸和Lewis酸作用下催化脱水形成中间体PA,最后由Lewis酸催化通过1,2-氢化物移位反应生成乳酸。研究表明,L-Nb2O5的高催化活性很可能是由于其具有大量的Lewis酸位点和Brønsted酸位点,这些位点来自于a、b轴的变形正交结构和c轴的额外添加层。其他无碱条件下催化甘油转化的反应机理也与上述过程相似[28,29]。
由于甲醇不可避免地存在于生物柴油生产的粗甘油中,理论上,用“一锅法”直接将甘油转化为乳酸烷基酯(乳酸甲酯)是简单易行的,并且不需要额外的分离以及酯化步骤。目前,甘油在甲醇或乙醇中直接转化为乳酸甲酯或乳酸乙酯的研究已成为热点。与甘油在水溶液中转化为乳酸的反应路径类似,甘油转化为乳酸甲酯包括3个连续反 应[16,30]:(1)甘油氧化生成DHA和GLA;(2)DHA和GLA脱水生成丙酮醛;(3)丙酮醛加成和异构化生成乳酸甲酯。Lu等[16]开发了Au/Sn-USY双功能催化剂,用于甲醇溶液中甘油一锅法转化为乳酸甲酯。一方面,Au与骨架外SnOx在DeAl-USY介孔周围的相互作用促进了Au颗粒的分散,有利于甘油氧化成DHA和GLA。另一方面,骨架Sn改变了催化剂的Lewis酸性质,使其具有独特的羰基活化能力,这对乳酸甲酯的生成至关重要。
2.3 甘油电催化氧化制乳酸
与传统的热催化氧化相比,电催化氧化不仅能在温和条件下实现甘油选择性氧化制乳酸,还能与阴极发生耦合反应,生成氢气。
Lux等[34]报道了将粗甘油电催化氧化为DHA和GLA,再进行碱催化将DHA或GLA转化为乳酸。Dai等[35]制备了AuPt双金属纳米催化剂,在常温常压下由甘油电催化氧化生产乳酸,反应路径如图4所示。甘油首先在AuPt催化剂和碱的作用下氧化成DHA或GLA,然后经过碱催化脱水生成2- 羟基丙烯醛或丙酮醛,最后经Cannizzaro重排生成乳酸。最近,Yan等[36]报道了一种光辅助电催化策略,在金纳米线(Au NWs)催化剂上实现了甘油选择性氧化制乳酸,乳酸选择性达80%。原位傅里叶红外光谱(FTIR)揭示了光照可以促进甘油在Au NWs表面的吸附,特别是甘油的仲羟基,这有助于产生DHA中间体,并通过碱催化进一步转化为乳酸,同时GLA中间体生成GLYA或C1~C2产物的副反应可以得到显著抑制。
目前电催化氧化法仍受限于电池的电流密度低等问题,阻碍了实际应用。另一方面,低成本、高选择性和高稳定性的电催化剂的开发也是电催化氧化面临的另一个主要挑战。
3 贵金属催化剂
3.1 Au基催化剂
表1 近年来Au基催化剂催化甘油氧化制乳酸的典型报道结果Table 1 Typical results for glycerol to lactic acid over Au-containing catalysts reported in the last years |
| Catalyst | Base a | T / ℃ | PO2 / MPa | Reaction time / h | Conversion / % | Selectivity b / % | Year | Ref |
|---|---|---|---|---|---|---|---|---|
| Au-Pt/TiO2 (1∶1) c | 4∶1 | 90 | 0.1 | - | ≈30.0 | LA 85.6 | 2010 | 15 |
| Au-Pt/TiO2 (3∶1) c | 4∶1 | 90 | 0.1 | - | ≈30.0 | LA 84.3 | ||
| Au-Pt/TiO2 (1∶3) c | 4∶1 | 90 | 0.1 | - | ≈30.0 | LA 85.3 | ||
| 1% Au/CeO2 d | 4∶1 | 90 | 0.1 | - | 99.1 | LA 73.1 | 2013 | 23 |
| 1% Au/CeO2 e | 4∶1 | 90 | 0.1 | - | 98.0 | LA 83.0 | ||
| 3% Au/CeO2 d | 4∶1 | 90 | 0.1 | - | 98.0 | LA 73.5 | ||
| 3% Au/CeO2 e | 4∶1 | 90 | 0.1 | - | 98.0 | LA 79.7 | ||
| 5% Au/CeO2 d | 4∶1 | 90 | 0.1 | - | 98.0 | LA 72.2 | ||
| 5% Au/CeO2 e | 4∶1 | 90 | 0.1 | - | 98.0 | LA 79.0 | ||
| Au/TiO2 | base free | 160 | 1 | 2 | 1.2 | - | 2013 | 24 |
| Pd/TiO2 | base free | 160 | 1 | 2 | 46.2 | LA 47.7 | ||
| AuPd/TiO2 | base free | 160 | 1 | 2 | 29.7 | LA 58.5 | ||
| 0.5Au/Sn-MCM-41-XS DP | base free | 140 | 3 | 4.5 | 76.0 | MLA 46.0 | 2018 | 51 |
| 0.5Au/Sn-MCM-41-XS CI | base free | 140 | 3 | 4.5 | 20.0 | MLA 82.0 | ||
| 0.5Au/CuO+Sn-MCM-41-XS | base free | 140 | 3 | 4.5 | 79.0 | MLA 64.0 | ||
| AuPd/CNTs | base free | 140 | 3 | 4.5 | 6.8 | MLA 5.7 | 2019 | 52 |
| AuPd/CNTs-NS+Sn-MCM-41-XS | base free | 140 | 3 | 4.5 | 81.0 | MLA 87.0 | ||
| AuPd/CNTs-NS+Sn-MCM-41-XS | base free | 140 | 3 | 9 | 96.0 | MLA 88.0 |
a NaOH to glycerol mole ratio if not otherwise denoted; b LA: lactic acid, MLA: methyl lactate; c Au to Pt mole ratio; d H2 reduced samples; e Glycerol reduced samples. |
在甘油的水相氧化体系中,碱的存在能够提 高Au基催化剂的活性[46]。催化剂表面结合氢氧化物,一方面可以显著降低Au(111)晶面的活化能[40],另一方面能够促进催化剂表面形成的醛或酮中间体发生β-氢化物消除反应,生成相应羧酸。Demirel-Gülen等[11]发现Au催化剂仅在碱性条件下可以有效催化甘油氧化。随着碱含量的增加,甘油氧化在动力学上更有利,说明Au催化剂需要碱的辅助才能实现甘油的氧化反应。Redina等[47]发现,当NaOH与甘油比例为2时,甘油对乳酸的转化率和选择性达到最佳,而NaOH与甘油比例过高容易导致碳酸盐大量生成,引起Au催化剂失活。Zope等[48]用18O2和H218O同位素标记实验证明,甘油氧化反应中,羟基氧化成羰基或羧基的氧来自氢氧根离子,而不是分子氧的氧原子。密度泛函理论(DFT)计算进一步表明,分子氧参与催化循环并不是通过金属位解离成原子氧,而是通过生成H2O2中间体催化分解形成氢氧根离子。除了碱度,提高反应温度也能提高Au基催化剂上乳酸的选择性。Purushothaman等[49]考察反应温度对甘油氧化制乳酸的影响,发现温度从140 ℃升高到180 ℃时,甘油的转化率从70%提高到80%左右,而乳酸的选 择性从24%提高到60%,说明高温有利于乳酸的生成。Xu等[24]在酸性条件下同样发现,甘油氧化生成乳酸需要较高的温度。甘油在100 ℃时转化率很低,并且没有乳酸的生成。当温度升高到140 ℃时,乳酸的选择性为57.5%。当温度进一步升高到160 ℃时,乳酸的选择性略有提高,而甘油转化率显著提高。
甘油分子在Au基催化剂上的氧化反应被认为是结构敏感反应,Au纳米颗粒粒径是控制甘油选择性氧化的重要因素。Bianchi等[45]发现Au/C催化剂在Au颗粒平均粒径为7~8 nm时,活性达到最大值。Lakshmanan等[23]发现乳酸的形成与Au颗粒尺寸密切相关。当Au/CeO2催化剂中Au含量为1%时,随着Au颗粒增大到~6 nm,对乳酸的选择性达到最大值;而Au/CeO2催化剂中Au含量为3%和5%时,中等大小的Au颗粒(~8 nm)对乳酸表现出最高的选择性,而较小或较大的Au颗粒均表现出较差的乳酸选择性。可见,在甘油氧化制乳酸反应中,最优的Au纳米颗粒尺寸应该在5~10 nm,此时乳酸选择性高。但是,大量使用Au纳米颗粒的涉氢和涉氧反应报道显示,Au纳米颗粒具备优异活性和选择性的尺寸范围应该是在更小的尺度范围(1~5 nm)。这种区别,与甘油转化制乳酸过程中,DHA和GLA(图3)生成和转化路径有关:小尺度的Au纳米颗粒有更有益的氧化脱氢能力,能够快速将GLA氧化脱氢生成GLYA,相应地,GLA异构生成DHA的路径被抑制。
合金化是提高Au基催化剂甘油氧化制乳酸活性和选择性的另一有效策略。在有碱存在的氧化体系中,Purushothaman等[31]比较了单金属Au/nCeO2催 化剂和双金属Au-Pt/nCeO2催化剂对反应性能的影响。单金属Au/nCeO2 TOF约为1100 mol·mol-1·h-1,乳酸选择性为60%。双金属Au-Pt/nCeO2催化剂,具有更高的TOF(1350 mol·mol-1·h-1)和乳酸选择性(80%)。需要注意的是,Pt金属在适宜Au催化剂的碱性体系下,活性和选择性较差。但是,通过将其与Au合金化,生成双金属Au-Pt,活性和选择性都得到改进。这种促进作用,与金属合金化的协同作用有关,还受金属-载体强相互作用的影响。研究中采用可还原性的CeO2与Pt金属耦合,适宜产生金属-载体强相互作用的效应:催化剂在还原处理或活化过程中,载体和金属界面处的CeO2发生部分还原,形成Ce3+和缺氧位点,同时部分Ce物种可以迁移覆盖到金属表面,抑制金属烧结。另外,由于金 属和载体之间的强相互作用,Ce4+离子同样改 变Pt和Au金属的化学价态,Au0表面活性位减少,增加了Au3+和Au+等新表面活性物种,有利于促进C—O和C=O等极性键的活化。Shen等[50]对比不同Au/Pt原子比(1/3~7/1)的Au-Pt/TiO2催化剂的甘油氧化性能,发现Au/Pt原子比为3∶1时活性最高(TOF为524 h-1)。该促进作用进一步说明,通过合金化,利用Au向Pt的电子转移增加了Pt的电子密度,改变表面Au和Pt活性金属的化学价态,能够促进了甘油仲羟基的氧化,促使DHA的形成优于甘油醛。Au和Pt之间的协同效应表明,通过合理调整金属催化剂的电子性质,可以有效地将甘油氧化为乳酸。在无添加碱的反应体系中,Xu等[24]发现AuPd/TiO2催化剂与AlCl3共同作用可以在无碱条件下选择性地将甘油直接氧化为乳酸,Au-Pd合金能将乳酸选择性从47.7%提高到58.5%。X射线光电子能谱(XPS)和透射电子显微镜(TEM)表征结果证明,Au和Pd之间存在相互作用。
Au催化剂同样在以甲醇为溶剂的甘油反应体系具有优异的性能。Purushothaman等[30]以Au/USY作为催化剂,甘油转化率为95%时,乳酸甲酯的收率为73%。Tang等[51]用金属氧化物负载Au纳米颗粒和Sn-MCM-41物理混合组成多功能催化体系,发现Au/CuO和Sn-MCM-41物理混合性能最佳,甘油转化率为95%,乳酸甲酯的收率63%。机理研究表明,Au基催化剂促进了甘油脱氢步骤,将甘油转化为DHA和GLA,固体酸Sn-MCM-41具有强Lewis酸和弱Brønsted酸将中间体DHA异构化为乳酸甲酯。该研究组进一步研究发现[52],碳纳米管(CNT)负载Au-Pd双金属和Sn-MCM-41物理混合组成多功能催化体系可将甘油转化率提高到96%,乳酸甲酯的收率高达85%。Zhou等[53]将Au/CuO和Sn-Beta物理混合组成双功能催化剂,实现了甘油向乳酸甲酯的高效转化。在90 ℃的低温条件下,可获得86%的甘油转化率和60%的乳酸甲酯收率。其中,Au/CuO为甘油氧化脱氢为DHA提供了氧化活性位点,而Sn-Beta为DHA进一步转化成乳酸甲酯提供了Lewis酸位点。Lu等[16]考察了反应温度、反应时间等条件对Au/Sn-DeAl-USY催化性能的影响。发现在一定的温度范围(100~140 ℃)内,甘油转化率和乳酸甲酯收率均逐渐增加,而较高的反应温度(>180 ℃)会导致过度氧化产物的增多。同样,根据不同反应时间的产物分布规律,可以有效推断反应的历程。
相较于Pt、Pd贵金属催化剂,Au吸附和活化氧气的能力较弱,因此其反应活性受体系酸碱性影响较为显著。而且,金属Au容易发生烧结或流失,如何通过载体或助剂引入,构筑多功能活性位,提高Au的活性和稳定性是当前Au基催化剂研究的 重点。
3.2 Pt基催化剂
Pt基催化剂在甘油氧化反应中面临的主要挑战在于提高抗氧化中毒能力和羟基定向活化。表2为部分Pt基催化剂催化甘油氧化制乳酸的研究结果。
表2 近年来Pt基催化剂催化甘油氧化制乳酸的典型报道结果Table 2 Typical results for glycerol to lactic acid over Pt-containing catalysts reported in the last years |
| Catalyst | Base a | T / ℃ | PO2 / MPa | Reaction time / h | Conversion / % | Selectivity b / % | Year | Ref |
|---|---|---|---|---|---|---|---|---|
| Pt/Al2O3 | 0.75∶1 | 240 | 3.5 | 22~30 | 95.4 | LA 29.8 | 2018 | 54 |
| Pt/ZnO | 0.75∶1 | 240 | 3.5 | 22~30 | 97.0 | LA 25.5 | ||
| Pt/MgO | 0.75∶1 | 240 | 3.5 | 22~30 | 93.6 | LA 30.8 | ||
| Au/nCeO2 | 4∶1 | 100 | 0.5 | 0.5 | 82.0 | LA 68.0 | 2014 | 31 |
| Pt/nCeO2 | 4∶1 | 100 | 0.5 | 0.5 | 60.0 | LA 52.0 | ||
| Au-Pt/nCeO2 | 4∶1 | 100 | 0.5 | 0.5 | 99.0 | LA 80.0 | ||
| 0.1%Cu-1.0%Pt/AC | 1.5∶1 | 90 | 0.1 | 4 | 43.7 | LA 66.9 | 2017 | 58 |
| 0.2%Cu-1.0%Pt/AC | 1.5∶1 | 90 | 0.1 | 4 | 52.7 | LA 65.7 | ||
| 0.35%Cu-1.0%Pt/AC | 1.5∶1 | 90 | 0.1 | 4 | 68.2 | LA 72.9 | ||
| 0.5%Cu-1.0%Pt/AC | 1.5∶1 | 90 | 0.1 | 4 | 80.0 | LA 69.3 | ||
| 0.75%Cu-1.0%Pt/AC | 1.5∶1 | 90 | 0.1 | 4 | 62.7 | LA 67.5 | ||
| 1.0%Cu-1.0%Pt/AC | 1.5∶1 | 90 | 0.1 | 4 | 60.9 | LA 64.4 | ||
| 2.0%Cu-1.0%Pt/AC | 1.5∶1 | 90 | 0.1 | 4 | 42.8 | LA 49.0 | ||
| Pt-Co/CeOx | 1.0∶1 | 200 | 1(N2) | 4 | 85.0 | LA 88.0 | 2019 | 65 |
| Pt/TiO2 | 4∶1 | 90 | 0.1 | 2 | 33.9 | LA 51.5 | 2014 | 66 |
| Pd/TiO2 | 4∶1 | 90 | 0.1 | 2 | 26.3 | LA 51.7 | ||
| Pd1Ni1Ox/TiO2 | 4∶1 | 90 | 0.1 | 2 | 44.0 | LA 51.6 | ||
| Pt1Ni1Ox/TiO2 | 4∶1 | 90 | 0.1 | 2 | 58.0 | LA 73.7 | ||
| Pt1Ni1Ox/TiO2 | 4∶1 | 90 | 0.1 | 4 | 99.1 | LA 62.6 | ||
| Pt/Sn-MFI | base-free | 100 | 0.62 | 24 | 89.8 | LA 80.5 | 2014 | 25 |
| Pt/Sn-BEA | base-free | 100 | 0.62 | 24 | 93.4 | LA 28.1 | ||
| Pt/silicalite-1 | base-free | 100 | 0.62 | 24 | 83.8 | LA 0.0 | ||
| Pt/AC + Sn-MFI | base-free | 100 | 0.62 | 24 | 53.6 | LA 80.8 | ||
| Pt/TiO2 | base-free | 100 | 0.62 | 24 | 92.3 | LA 0.0 | ||
| 0.1Pt/L-Nb2O5 | base-free | 140 | 0.5 | 3 | 9.0 | LA 92.0 | 2020 | 27 |
| 0.5Pt/L-Nb2O5 | base-free | 140 | 0.5 | 3 | 53.0 | LA 79.0 | ||
| 1Pt/L-Nb2O5 | base-free | 140 | 0.5 | 3 | 69.0 | LA 61.0 | ||
| 2Pt/L-Nb2O5 | base-free | 140 | 0.5 | 3 | 84.0 | LA 28.0 |
a NaOH to glycerol ratio if not otherwise denoted; b LA: lactic acid. |
载体对Pt基催化剂的性能影响显著。Bruno 等[54]采用湿浸渍法制备了不同氧化物(Al2O3、ZnO、MgO)负载的Pt催化剂,发现Pt/Al2O3、Pt/ZnO、Pt/MgO催化剂对乳酸的选择性分别为45%、60%和55%。Pt/Al2O3催化剂对乳酸的选择性最低,通常被认为是由于其碱性密度较低(0.3 μmol CO2 m-2)导致的。但是,与碱性载体MgO相比,Pt/ZnO催化剂的碱度(35.8 μmol CO2 m-2)低于Pt/MgO催化剂(60.9 μmol CO2 m-2),但Pt/ZnO催化剂对乳酸的选择性和收率优于Pt/MgO。这充分说明,乳酸的选择性不完全依赖于载体酸碱性,还与载体的结构有关。ZnO作为n型半导体,早期是合成气制甲醇的活性组分,说明其对于活化C—O或C=O这类极性键具有优异的性能。Pt/ZnO催化剂,ZnO不仅可以作为载体支撑Pt高分散,同时Zn2+本身具备Lewis酸特性,可以活化DHA中的C—O键,促进DHA向PA的转化。这一点与Ftouni等[55]比较Pt/C、Pt/TiO2、Pt/ZrO2三种催化剂的甘油氧化性能的结果相互印证。Pt/C催化剂的乳酸选择性为60%,而采用n型半导体TiO2或ZrO2的催化剂,乳酸选择性超过80%。Checa等[56]进一步对比了不同可还原金属氧化物(TiO2、ZnO、SnO2和ZrO2)载 体负载Pt催化剂的甘油转化性能。结果发现,以 ZnO和SnO2为载体的Pt催化剂表现出最高的氧化活性,原因是金属和载体界面处的Zn或Sn原子,因为发生金属-载体强相互作用,生成了Pt-Zn或Pt-Sn合金结构,促进了甘油转化。Komanoya等[28]证明,Pt纳米颗粒和TiO2组合催化剂可以在氧气气氛下实现甘油到乳酸的转化。即使在没有碱的情况下,乳酸的收率也能达到63%。其中,TiO2上的Lewis酸位 点促进了中间体的脱水和重排,从而高效地生产乳酸。Wu等[57]发现载体本身可以参与反应,提高催化剂的活性,影响产物的选择性。采用溶胶凝胶法制备 了Pt/CuO、Pt/TiO2和Pt-Sol催化剂用于甘油氧化反应,结合原位红外光谱技术和反应动力学研究结果,发现Pt/CuO和Pt/TiO2催化剂表现出不同的反应路径(图5)。Pt/CuO催化剂表面O原子通过氢键与甘油仲羟基产生强相互作用(β相互作用),在反应过程中通过降低甘油仲羟基的C—O键电子云密度实现对甘油仲羟基定向氧化。而Pt/TiO2催化剂表面Ti原子通过与甘油伯羟基形成醇盐,产生强的金属氧相互作用(γ相互作用),反应时甘油伯羟基C—O键电子云密度降低,从而提升甘油伯羟基氧化的选择性。
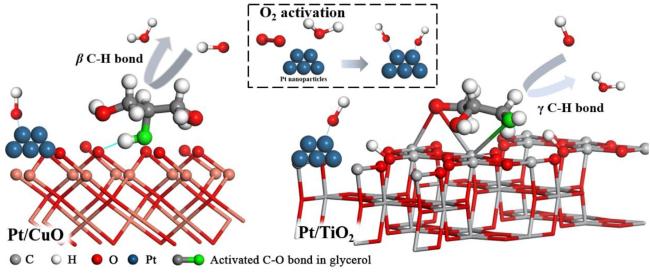
总之,将可还原性的n型半导体氧化物载体与金属Pt耦合,可利用金属-载体强相互作用提高金属Pt的热稳定性和抗烧结稳定性,同时利用界面反应生成新的活性相,可显著改善甘油转化的活性和选择性。
除了载体效应,Pt金属纳米颗粒的结构和尺寸也是影响催化活性的重要因素。Zhang等[58]通过改变沉淀-沉积温度(0~80 ℃)制备了一系列Pt粒径范围在10.2~3.8 nm的Pt/AC催化剂,研究了Pt/AC催化剂在碱溶液中催化甘油制乳酸反应中Pt颗粒的尺寸效应。其中,中等大小Pt颗粒(平均粒径 为7.9 nm)Pt/AC-30催化剂的TOF值最高,甘 油转化率为64.1%时,乳酸选择性达42.9%。但是,乳酸选择性受Pt粒径效应影响不明显。此外,粒径效应还与碱类型有关,与在KOH溶液中相比,Pt金属粒径尺寸对甘油转化率的影响在LiOH溶液中更为明显。Oberhauser等[59]制备了1.5 nm的Pt纳米颗粒,并将其负载到碳材料上,所得Pt@C展现出优越的催化活性(780 h-1)和乳酸选择性(94%)。
甘油的端羟基在金属Pt催化剂上优先被活化,因此Pt催化剂用于甘油氧化反应,容易生成甘油 酸[60⇓⇓~63]。为提高乳酸选择性,通常采用向Pt金属中引入第二种金属形成合金或双金属催化剂。Zhang等[64]将Cu引入到Pt/AC催化剂中,发现Cu引入提高了Pt分散。Cu与Pt之间的强相互作用提高了甘油氧化活性,0.5%Cu-1.0%Pt/AC催化剂的乳酸收率是1.0% Pt/AC催化剂的2.4倍。进一步的研究表明,Pt/Cu比不同,催化剂Cu物种的价态发生变化。其中Cu+和Cu0物种有利于甘油活化和乳酸生成,而Cu2+(大的CuO颗粒)则有利于甘油酸的形成。Zhang 等[65]制备了双金属Pt-Co/CeOx催化剂,发现电子从Co0转移到Pt0,同时Pt-Ce发生相互作用。甘油和NaOH都需要在Pt-Co表面被活化,Pt-Co之间独特的电子耦合效应和Co掺入引起的CeOx晶格畸变使得催化剂具有优异的性能。200 ℃反应条件下甘油氧化制乳酸的活性(1533.9 h-1)和选择性(87.7%)显著提高。Li等[66]制备了Ni促进的PtmNinOx/TiO2催化剂,研究发现,与Pt/TiO2相比,NiOx与金属Pt协同催化甘油氧化,从而显著提高催化剂的甘油转化活性,甘油转化率为99.1%时,乳酸的选择性为62.6%。
Pt基催化剂具有优异的氧化脱氢性能,金属颗粒和载体大小是影响反应过程的关键因素。虽然Pt基催化剂在甘油氧化中表现出较好的催化活性和选择性,但是稳定性较差,容易发生氧中毒和金属浸出。通过引入第二种金属对催化剂改性,可以形成合金以及增加活性位点数量,不仅可以提高反应活性,催化剂稳定性也大大提高。
3.3 其他贵金属催化剂
除了Au和Pt催化剂,Pd、Ru和Ag催化剂也被用于甘油氧化。Pd催化剂对甘油酸的选择性更高[67],且需要在反应体系中添加碱提高活性。Marques等[68]将Pd/AC催化剂用于甘油氧化制乳酸的反应,在230 ℃、NaOH/甘油摩尔比为1.1的条件下,甘油转化率为99%,乳酸的选择性为46%。Arcanjo等[69]研究了活性炭负载Pd催化剂的活性,催化剂金属负载量、NaOH/甘油摩尔比、温度等参数对催化性能有显著影响。在230 ℃的反应温度下,10% Pd/C催化剂的甘油转化率可达约99%,乳酸的选择性为68%,但Pd 负载量高。Shen等[70]以羟基磷灰石(HAP)负载3%Pd制备Pd3/HAP催化剂,在NaOH/甘油摩尔比为1.1,反应温度为230 ℃条件下,甘油转化率为99%时,乳酸选择性高达95%。通过离心将Pd3/HAP催化剂从溶液中分离出来,进行5次重复实验后,甘油转化率从99%下降到93%,乳酸选择性从95%下降到94%。Pd3/HAP催化剂的催化活性和选择性略有下降,是 由于微量Pd从载体表面浸出到溶液中。最近,Ten等[71]制备了双金属Pd,Bi@Uio-66催化剂用于甘油级联转化为乳酸的反应,混合材料的独特结构和可调性使得在不添加任何碱的情况下,甘油转化率为19%时,乳酸的选择性为58%。由此可见,Pd催化剂在无碱条件下很难获得高活性。
Ru具有较高的氧化和配位数,因此Ru金属配合物通常具有良好的氧化反应活性。Li等[72]提出了一种利用钳形Ru配合物从甘油制氢和选择性合成乳酸的方法。在125 ℃下,使用少于1 ppm的Ru-MACHO催化剂反应2 h,获得了前所未有的TOF值,乳酸的收率为67%。此外,对甘油脱氢产物的分析表明,提高脱羧效率是进一步重整工艺的关键。值得注意的是,使用Ru基催化剂从甘油中生产乳酸存在一个问题,Ru容易使碳氢化合物中的C—C键断裂。因此,在碱性条件下,Ru纳米颗粒催化甘油转化为乳酸时,乳酸分解为甲酸不可避 免地会形成甲烷和二氧化碳[73]。为了解决上述难题,Jiang等[74]报道了一种羟基磷灰石(HAP)负载三金属Ru-Zn-CuI/HAP催化剂将甘油转化为乳酸。结果表明,Ru颗粒(< 2 nm)均匀分散在HAP上。CuI能有效抑制C—C键的裂解,乳酸的选择性显著提高。同时,Zn2+的存在提高了中间产物丙酮醛的异构化。与Ru/HAP催化剂(63.7%)和Ru-Zn/HAP催化剂(70.9%)相比,Ru-Zn-CuI/HAP催化剂对乳酸选择性提高到82.7%。Pemmana等[75]研究了活性炭负载Ru-V双金属催化剂在甘油转化制乳酸中的应用。Ru/V2O5(Ru-V/AC)双金属催化剂性能优于单金属Ru和V催化剂,在温和的反应条件下,Ru-V/AC双金属催化剂表现出最佳的催化性能,甘油转化率为98.7%时,乳酸收率为75.5%。
与其他贵金属不同,Ag基催化剂的甘油氧化活性最弱。Lari等[76]提出了一种以丙酮醛为中间体的甘油制乳酸转化工艺,即甘油气相氧化水合制丙酮醛,丙酮醛连续液相转化制乳酸或乳酸甲酯。Tao等[77]制备了银交换的磷钼酸催化剂AgxPMo12O40 (x=1,2,3),并对其甘油转化制乳酸性能进行了评价。结果表明,Ag金属的添加可以提高H3PMo12O40的氧化还原电位,使其有利于将甘油转化为DHA而不是GLA,而Ag的Lewis酸位点有利于DHA进一步脱水转化为乳酸。适宜的氧化还原电位、Lewis酸位点和疏水性使其具有高转化率和高选择性。在不添加任何碱的情况下,甘油转化率为99%时,Ag3PMo12O40对乳酸的选择性达到了93%。同时,Ag3PMo12O40作为多相催化剂进行12次循环反应后而保持高活性。
贵金属的高成本和有限的可重复利用性,使得开发低成本的甘油转化催化剂成为未来极具吸引力的方向。因此,将非贵金属活性相引入催化剂是未来催化剂设计的重点。
4 催化剂载体选择及作用
如前所述,载体对催化剂性能的影响已被多次提及。载体可以影响贵金属的结构、尺寸和电子性质,同时还可以引入新的功能位点(如酸性或碱性位点等)影响反应物活化。目前,甘油氧化制乳酸催化剂载体主要有碳材料、分子筛和金属氧化物。
4.1 碳材料
碳材料比表面积大、导电性好、耐酸碱,且通过高温燃烧可实现贵金属的回收,因此常用于负载贵金属催化剂[78]。
活性碳(AC)是具有较强吸附能力碳材料的总称,活性碳的比表面积和孔径分布都会影响催化剂的活性。Zhang等[79]研究了不同粒径AC负载Pt 催化剂在甘油选择性氧化制乳酸反应中的催化性能,发现Pt纳米颗粒高度分散在AC表面,平均粒径在2.8~5.0 nm,这些高度分散的Pt纳米催化剂在无碱条件下的甘油氧化反应中表现出较高的活性。AC粒径的减小使得Pt纳米颗粒对反应物可及性提高,进而反应活性提高。
碳材料的N掺杂可以引入表面官能团[80],包括吡啶N、吡咯N和石墨N,这些官能团能够锚定金属颗粒,起到改善金属分散性的作用。此外,掺杂的含N基团与金属纳米颗粒发生电子相互作用,从而产生新的催化行为。Chen等[81]发现在碳纳米管(CNTs)中掺入N原子会增加催化剂表面碱度,从而加速甘油羟基的活化。在无碱水溶液中,掺入N原子的Pt/N-CNTs催化剂对甘油氧化反应的活性远高于未掺N的Pt/CNTs催化剂。通过掺杂N而提高催化活性的原因是:(ⅰ) 电子从N转移到Pt;(ⅱ) 加速了分子氧的活化;(ⅲ) 表面缺陷位点的增加促进了甘油和O2的吸附;(ⅳ) 表面碱性的增加使甘油中的—OH基团更容易被活化。研究还指出,这些催化剂可以被重复使用多次,但催化剂会发生氧中毒(形成PtOx物种),需要在H2下进行再生。Zhang等[82]使用N掺杂的多壁碳纳米管(MWCNTs)作为Pt纳米颗粒的载体,在无碱条件下进行甘油氧化反应。结果与Chen等[81]结论一致,N的掺入有利于Pt物种的高分散,同时Pt纳米颗粒保持富电子态。
4.2 分子筛
含Sn分子筛中Lewis酸位点和Brønsted酸位点的结合可以极大地提高催化活性。Asgar等[86]合成了含L酸位点(骨架Sn)和B酸位点(骨架Al)的Sn-Al-Beta分子筛,并与AuPd/CNTs混合,作为甘油一锅法转化为乳酸甲酯的多功能催化体系。在140 ℃条件下,甘油转化率为29%,乳酸甲酯选择性为67%。
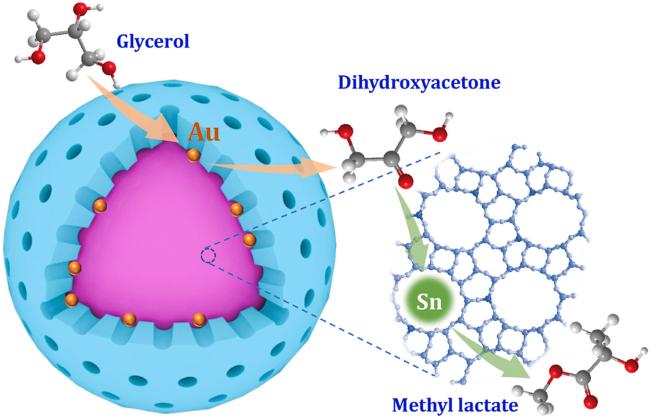
由于甘油分子尺寸较大,分子筛的晶内扩散限制也会影响催化效果。构建多级孔结构分子筛可显著改善扩散性能。Tang等[87]通过在微孔Snβ沸石上定向组装介孔二氧化硅,构筑了核壳结构的Snβ@介孔二氧化硅(MS)复合材料。壳层介孔通道与Snβ内核垂直对齐,实现了外壳与内核之间的充分传质。在此基础上,制备了Au/Snβ@MS催化剂(图6)。在最佳的反应条件下,1Au/Snβ-50@MS-1.5催化剂的活性显著提高,甘油转化率达到98.2%,乳酸甲酯选择性达到91.9%。介孔二氧化硅的引入可以有效改变Snβ@MS的L酸与B酸比例,还能促进高分散Au纳米颗粒的形成。此外,催化剂核壳结构可以有效地保护Au纳米颗粒免受烧结,从而表现出较高的稳定性和可回收性。Cho等[88]通过加入三维有序介孔碳模板剂成功合成了多级孔Sn-MFI分子筛,其介孔比例高。另外,硅羟基缺陷产生的弱酸中心能促进反应物活化。
4.3 金属氧化物
金属氧化物载体具有组成可调、形貌可控、与负载金属之间存在较强的相互作用等优点,可以抑制活性金属的团聚和流失、加速电子传递,从而增加催化剂活性和稳定性。另外,金属氧化物自身存在酸碱性、氧吸附活性等还可能起到助催化剂的作用。
在甘油氧化制乳酸的反应中,通常使用金属氧化物来提高路易斯酸强度,如TiO2、CuO、Al2O3、CeO2、ZnO和SnO2。但是,氧化物载体有限的表面积限制了贵金属的分散。为解决这个问题,Douthwaite等[89]以SBA-15作为硬模板剂,采用纳米铸造合成了一种高比表面积介孔TiO2材料(110 m2/g),进而制备了AuPt/TiO2催化剂。发现纳米铸造TiO2负载的AuPt催化剂展现出更高的甘油转化率(99%)和乳酸选择性(82%)。研究认为,SBA-15刻蚀后残留在TiO2表面大量的Si会形成B酸位点,而与NaOH作用会形成L酸位点,这两种位点均能促进甘油伯羟基的脱水,从而提高了乳酸的选择性。
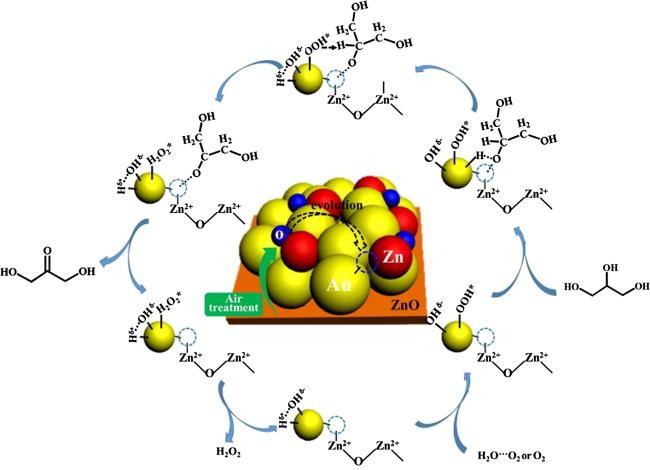
贵金属-可还原性氧化物体系存在金属-载体强相互作用(SMSI),可改变金属的几何和电子结 构[90]。Pan等[91]采用沉淀沉积法(DP)制备了Au/ZnO催化剂,并控制空气热处理条件(Au/ ZnODP-Air),在ZnO层和Au纳米颗粒之间产生强SMSI。结果表明,ZnO在空气热处理过程中迁移到Au纳米颗粒表面,形成Au-O-Zn界面以及大量的氧空位。甘油的仲羟基可以在生成的界面位点上被吸附活化,从而具有较高的DHA选择性(图7)。具体来说,甘油的仲羟基被解离吸附在Au/ZnO界面的氧空位缺陷上,形成吸附的(R2)CHO*和H*物种。因此,这种界面结构的制备是实现甘油选择性转化的有效策略。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
金属氧化物钙钛矿具有较高的结构耐受性,是研究载体化学组成变化如何影响催化剂性能的 理想材料。Evans等[92]以LaBO3钙钛矿(其中B=Cr,Mn,Fe,Co或Ni)为载体制备双金属AuPt纳米催化剂用于甘油氧化反应。通过改变B位点,催化剂的催化活性和选择性随着氧吸附能力的改变而发生显著变化。例如,AuPt/LaMnO3催化剂对甘油酸的选择性为70%,而在相同的反应条件下,AuPt/LaCrO3催化剂对乳酸的选择性为86%。
4.4 其他载体
LREHs是一种二维层状结构的新型稀土材料,由富含正电荷的稀土金属氢氧化物主体层板和层间客体阴离子构成[95]。目前,关于LREH材料用于催化领域的报道不多,有待进一步开发。Wang等[94]通过反微乳法制备了一系列均匀的LREH(RE=Y, La, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er和Tm)纳米片,再经过沉淀-沉积法负载Au催化剂(Au/LREO)。Au/ LREO的催化活性和结构表征表明,稀土离子的种类对Au的粒径、价态和还原性起关键作用,是影响Au在甘油转化过程中催化活性的重要因素。其中Au/LPrO表现出最好的性能,包括最高的甘油转化率(68.2%)、乳酸选择性(66.8%)和C3产物选择性(90.6%)。事实上,金颗粒的超小尺寸(~3 nm)、高Au0组分含量和高还原性是实现Au/LPrO优异催化性能的必要前提。
5 催化剂失活与再生
如前所述,贵金属催化剂适宜用于液相体系催化甘油选择性氧化制乳酸。但是,反应后的催化剂通常由于发生过度氧化、产物或副产物强吸附失活、金属浸出或金属烧结等问题导致催化剂无法重复使用[37,39,96]。报道较多的失活类型是过度氧化导致催化剂失活[62]。化学吸附的氧可以形成强M—O或M—OH键,部分覆盖金属表面,导致催化剂过度氧化。表面吸附氧的覆盖度取决于金属对氧和有机底物的相对亲和力,以及反应的pH、溶剂、温度和氧分压。这种动态氧化还原平衡控制着反应速率,并且随着底物浓度的降低,反应平衡向氧覆盖度高的方向移动,从而毒害催化剂[10]。氧化还原电位高的金属不易氧化,因此,在常见的贵金属催化剂中,抗氧化能力顺序为:Au>Pt>Pd。Au基催化剂可以在较高的氧气压力下反应,而不会出现催化剂失活的迹象。当使用Pt或Pd基催化剂时,必须使用较低的氧气压力甚至是大气压,以防止其过度氧化。除此之外,在酸性条件下,未解离的有机酸(如甘油酸),强烈吸附在催化剂的活性位上,增加了表面过氧化,更容易造成失活。
总的来说,开发用于甘油选择性氧化制乳酸的多相催化剂仍是一项极具挑战性的工作。在保证甘油转化高活性和乳酸高选择性的同时,需要重点开发可回收和可循环利用的催化剂。
6 结论与展望
目前,乳酸的生产主要依赖生物发酵法,但该方法存在苛刻的反应条件、环境污染严重、生产效率低下的问题。与生物发酵法相比,生物基甘油催化转化法具有反应条件温和、生产过程环保等优点。本文综述了贵金属催化剂甘油选择性氧化制乳酸的研究进展。首先,简要介绍了甘油转化制乳酸的反应机理,包括水热转化、选择性氧化和电催化氧化,值得一提的是,甘油在甲醇体系中的转化避免了水相反应体系需要相分离、提纯的繁琐;然后,重点从Au基催化剂、Pt基催化剂等贵金属催化剂对甘油选择性氧化制乳酸反应性能影响进行了介绍和评述,氧化脱氢是甘油催化转化为乳酸的关键步骤,贵金属及其合金在甘油脱氢反应中具有显著的活性;最后,从催化剂设计的角度出发,分析了载体种类和性质对催化剂活性的影响,并对催化剂的失活机理进行了讨论,而引入合适的功能位点是获得优异催化性能的关键。目前,虽然甘油选择性氧化制乳酸催化剂的设计与结构调控相关研究已经取得较大进展,但未来研究乃至面向工业应用仍面临以下几方面的机遇和挑战:
(1)甘油氧化反应网络复杂,聚焦“精准催化”的理论体系与技术方法,实现甘油定向活化是提高乳酸选择性的主要研究方向。具体而言,需要深 入探究金属活性位在甘油伯、仲羟基氧化中的作用。特别是贵金属催化剂,需要进一步明确金属-载体界面作用对羟基活化的影响,以确定最佳的金属种类和金属负载形式,针对性地改进、优化催化剂制备策略。此外,目前大多数高活性和高选择性催化剂仍以贵金属基催化剂为主,但工业应用成本极高。减少贵金属用量,将单原子或原子级分散的负载型金属催化剂的设计和合成理念引入到该体系催化剂中,制备贵金属单原子或双原子催化剂,有望在降低贵金属成本的同时进一步提高反应选择性。另外,设计开发非贵金属催化剂仍然是降低成本的最优选择。可以将类贵金属化合物(如氮、磷、碳等化合物)或高熵合金的理念引入过渡金属催化剂的开发,调变过渡金属活性低的问题,将是未来甘油高效转化催化剂研发的一条重要的发展方向。
(2)Lewis酸在甘油催化转化为乳酸的过程中起着重要作用,可以在无碱条件下实现乳酸的高收率。将四价Sn4+金属离子掺入二氧化硅骨架中是合成Lewis酸催化剂的有效策略,并表现出显著的活性和选择性。尽管如此,大规模使用这种材料仍然面临一些重要问题。从材料合成的角度来看,探究新的合成方法,增加骨架Sn物种的引入量以提高Lewis酸性,制备多级孔分子筛以提高传质和活性位的可及性,是有效利用分子筛进行生物质转化的重大挑战。目前,分子筛酸性和氧化还原性质表征通常依赖与活性位相互作用的探针分子的吸附,而针对溶剂介质中活性位的吸附研究是有限的,未来需要发展原位表征技术和方法,以更好地了解液相催化中活性位的行为。
(3)催化剂功能位点的精准构筑和活性位协同作用机制是生物质催化领域的研究前沿。深入解析构效关系,需要采用先进的原位表征技术与理论模拟计算,从分子、原子角度明确反应物与活性位点相互作用机制。此外,在液相反应体系中,贵金属催化剂常常由于金属浸出或中毒而失活。结合实验表征和理论计算,从微观角度深入分析活性位毒化过程和失活原因,以开发低成本、高选择性、高稳定性催化剂是今后研究的重要方向。