



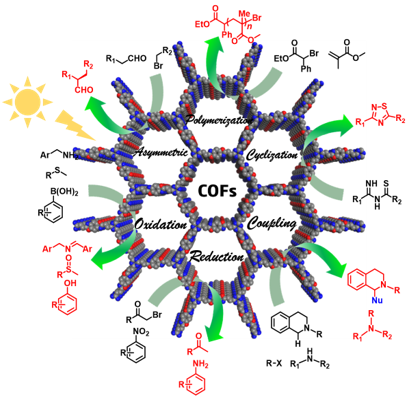
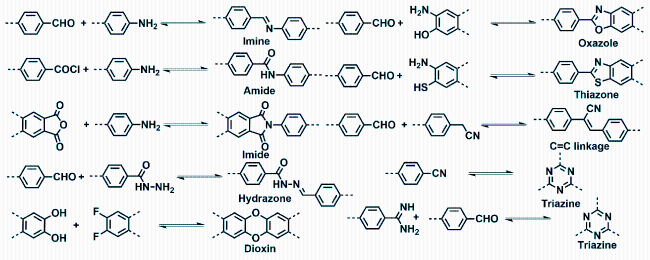
1 引言
 英、
英、  唑、噻唑、碳碳双键、三嗪等[29]。而COFs连接方式的发展趋势是由可逆共价键逐渐向不可逆共价键转化,这样不仅能够提高COFs的稳定性同时还能推动COFs的广泛应用。
唑、噻唑、碳碳双键、三嗪等[29]。而COFs连接方式的发展趋势是由可逆共价键逐渐向不可逆共价键转化,这样不仅能够提高COFs的稳定性同时还能推动COFs的广泛应用。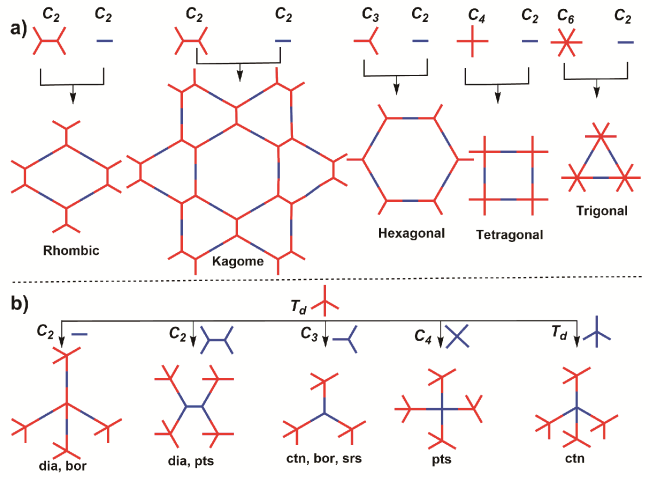
2 构建光功能化COFs的策略
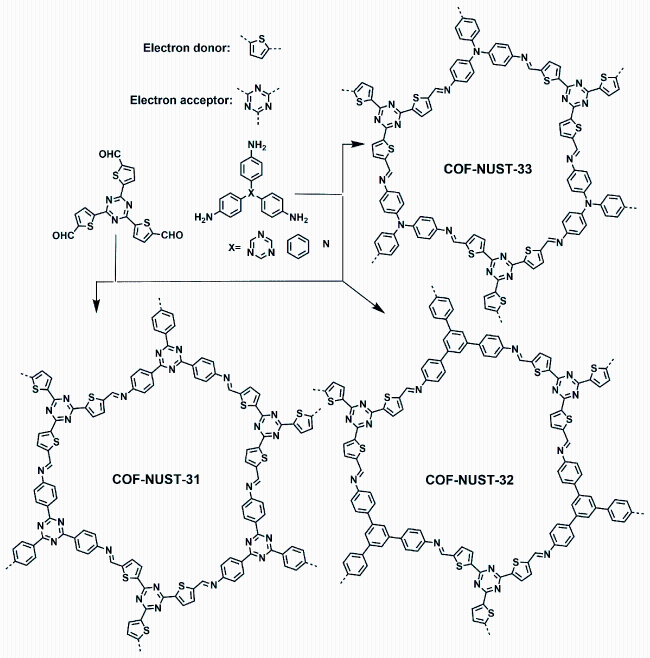
2.1 自下而上的策略
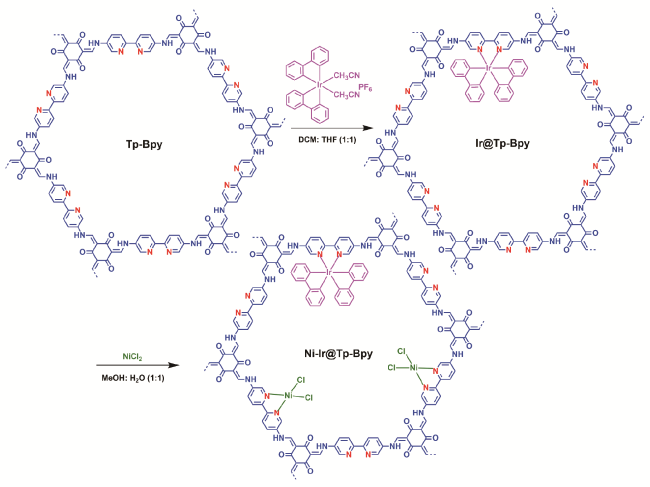
2.2 后修饰方法
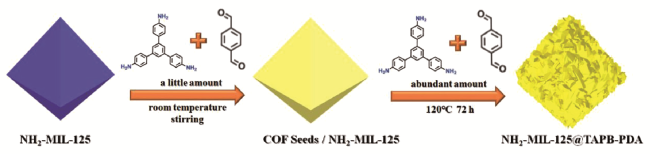
2.3 复合法
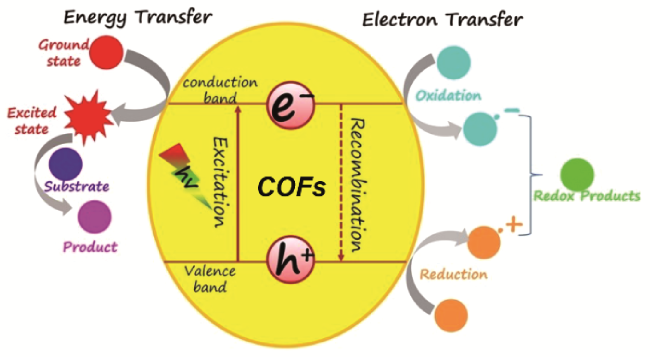
3 基于COFs光催化剂的光催化有机反应机理
4 COFs光催化的有机反应
4.1 氧化反应
4.1.1 伯胺氧化成亚胺
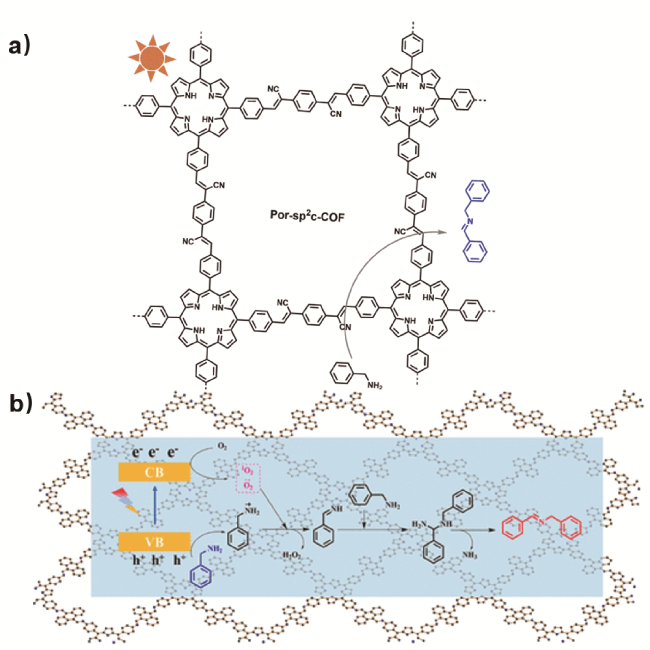
图7 a)Por-sp2c-COF光催化氧化胺成亚胺的示意图;b)Por-Ad-COF光催化氧化胺成亚胺的反应机理[60]Fig. 7 a) Por-sp2c-COF used as photocatalyst for the visible-light-induced aerobic oxidation of amines to imines. b) Plausible photocatalytic mechanism in the homocoupling of amines to imines over Por-Ad-COF[60]. Copyright 2021, American Chemical Society |

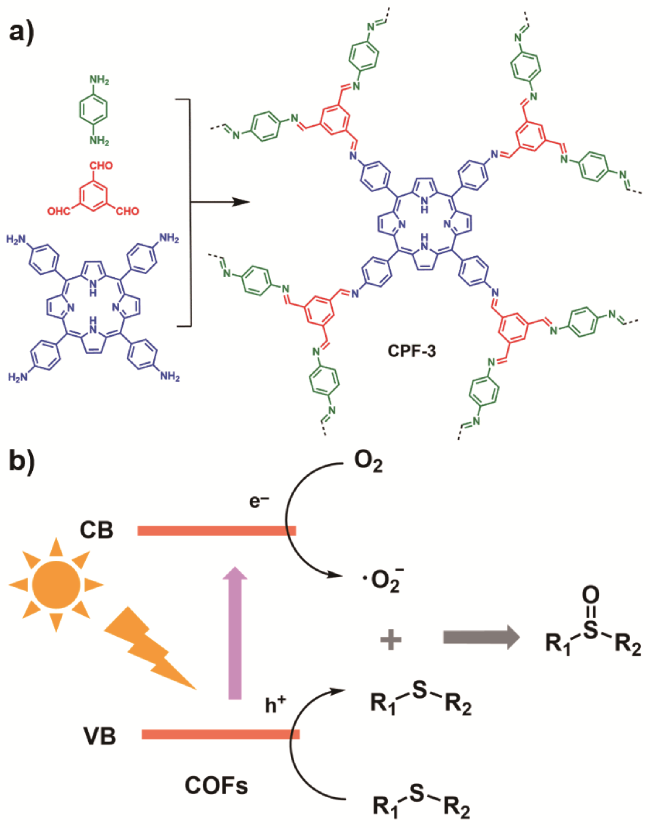
4.1.2 硫醚选择性氧化成亚砜
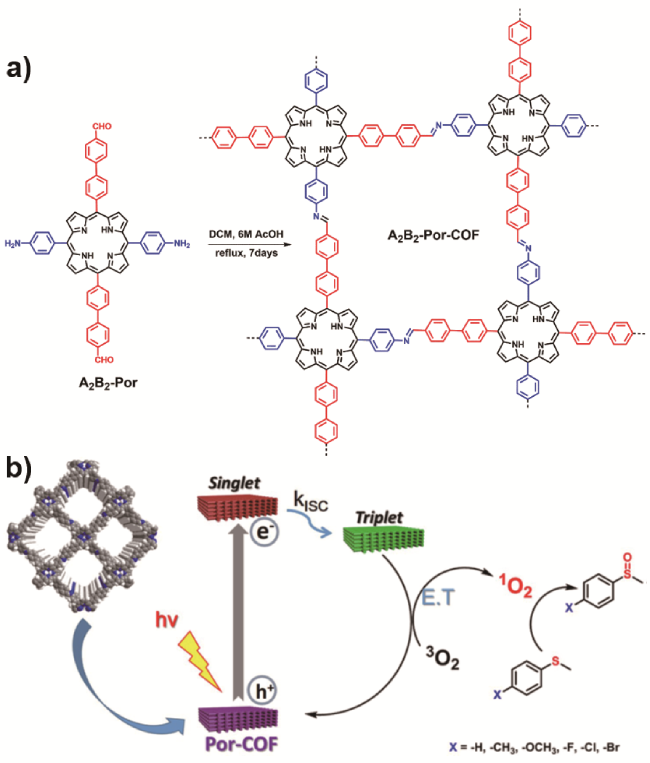
图9 a)由单体A2B2-Por自缩合构建A2B2-Por-COF结构示意图. b)A2B2-Por-COF光催化氧化硫醚成亚砜的反应机理[66]Fig. 9 a) Construction of A2B2-Por-COF by the self- condensation of A2B2-Por monomer. b) Proposed reaction mechanism for the photocatalytic oxidation of thioanisole by A2B2-Por-COF[66]. Copyright 2019, American Chemical Society |
4.1.3 苯硼酸氧化成苯酚
 唑连接的超稳定COFs(LZU-190、LZU-191和LZU-192),并将其作为无金属的多相光催化剂(图11a)促进芳基硼酸的光氧化。由于这些COFs是通过苯并
唑连接的超稳定COFs(LZU-190、LZU-191和LZU-192),并将其作为无金属的多相光催化剂(图11a)促进芳基硼酸的光氧化。由于这些COFs是通过苯并  唑而连接,它们在强酸、强碱等苛刻条件下均表现出超强的稳定性。同时,苯并
唑而连接,它们在强酸、强碱等苛刻条件下均表现出超强的稳定性。同时,苯并  唑的存在增强了COFs对可见光的吸收,缩小了带隙。例如,LZU-190作为光催化剂促进芳基硼酸的氧化羟化时,其产率高达99%,且催化循环20次后仍能保持高的催化活性。通过电子顺磁实验证明LZU-190是反应体系能够产生O2• −的关键,通过18O标记实验揭示了产物中的氧来源于O2。根据上述控制实验结果,他们提出了可能的光催化反应机理(图11b)。该研究为有机小分子框架化成刚性多相催化剂提供了思路,也为进一步研究其构-效关系作出了重要贡献。
唑的存在增强了COFs对可见光的吸收,缩小了带隙。例如,LZU-190作为光催化剂促进芳基硼酸的氧化羟化时,其产率高达99%,且催化循环20次后仍能保持高的催化活性。通过电子顺磁实验证明LZU-190是反应体系能够产生O2• −的关键,通过18O标记实验揭示了产物中的氧来源于O2。根据上述控制实验结果,他们提出了可能的光催化反应机理(图11b)。该研究为有机小分子框架化成刚性多相催化剂提供了思路,也为进一步研究其构-效关系作出了重要贡献。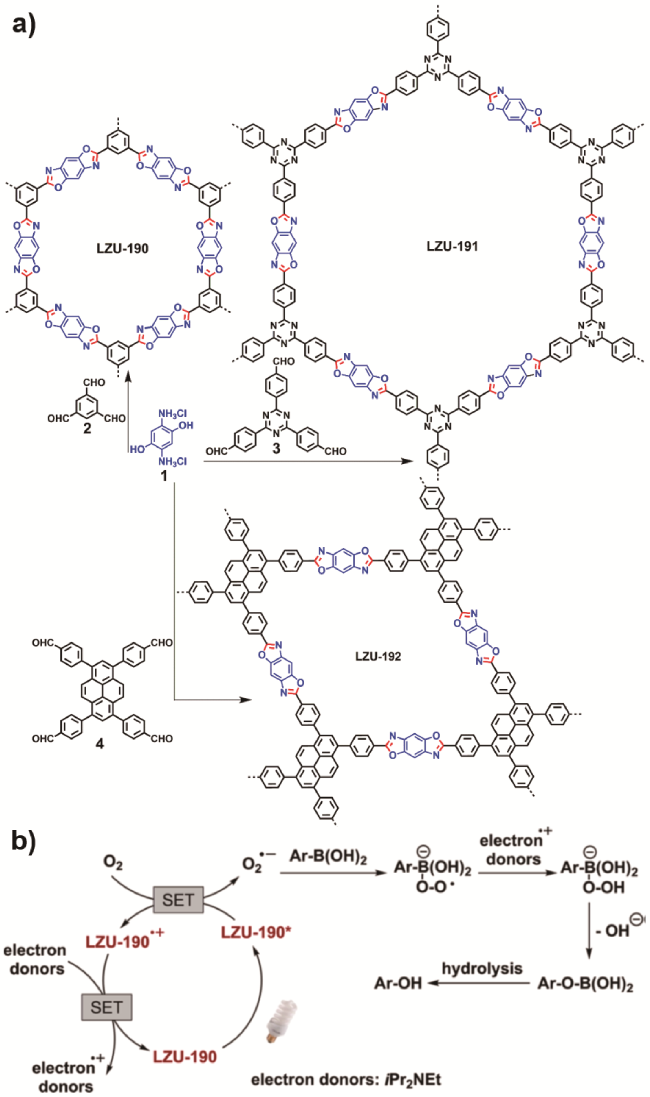
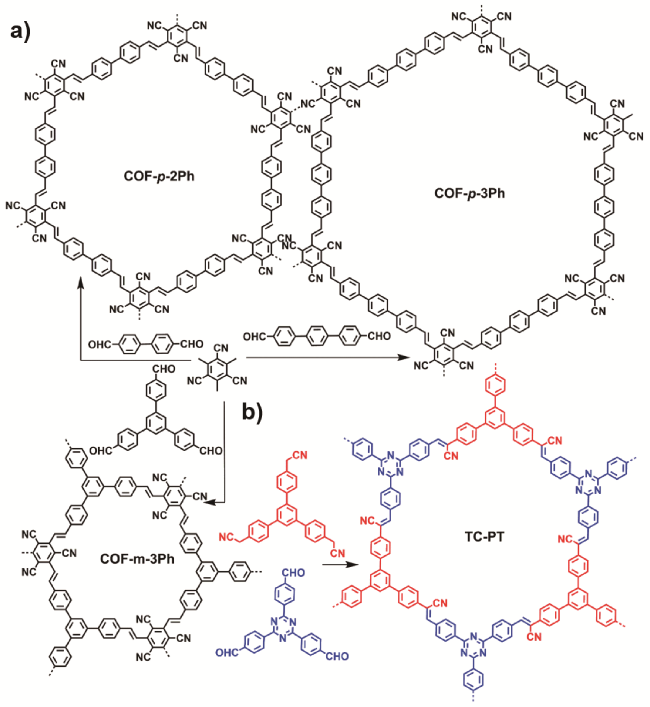
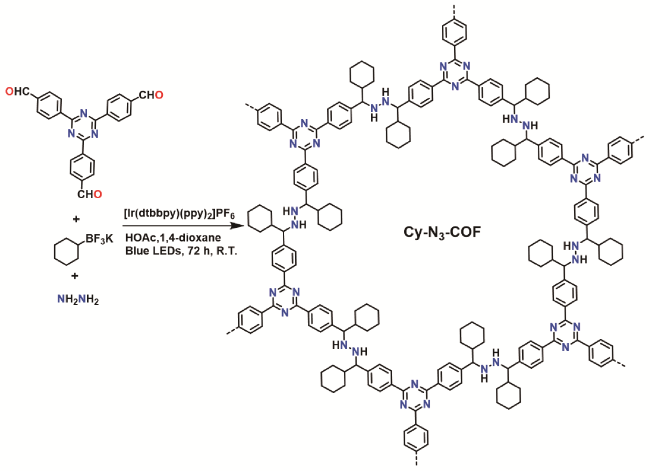
4.1.4 N-芳基四氢异喹啉的氧化
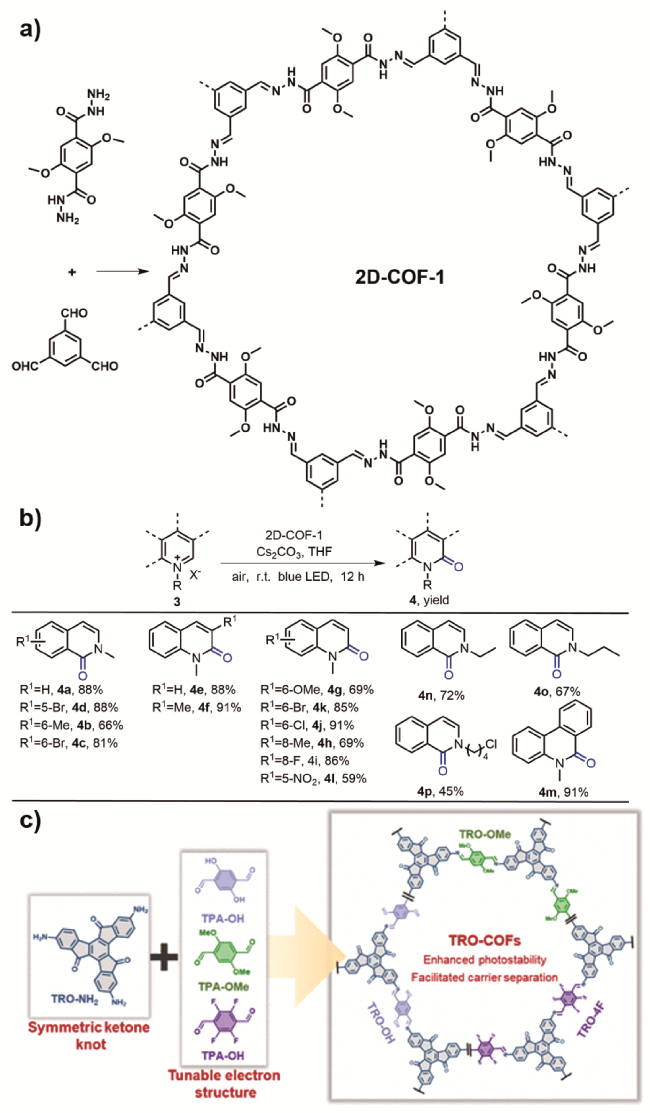
图14 a)2D-COF-1的合成[80];b)2D-COF-1作为光敏剂选择性氧化四氢异喹啉[80];c)TRO-COFs的合成[81]Fig. 14 a) Synthesis of 2D-COF-1[80]. b) Visible-light-driven selective oxidation of N-alkylpyridinium salts into quinolones by using 2D-COF-1 as the photosensitizer[80]. c) Synthesis of TRO-COFs[81]. Copyright 2023, American Chemical Society |
4.2 还原反应
4.2.1 脱卤反应
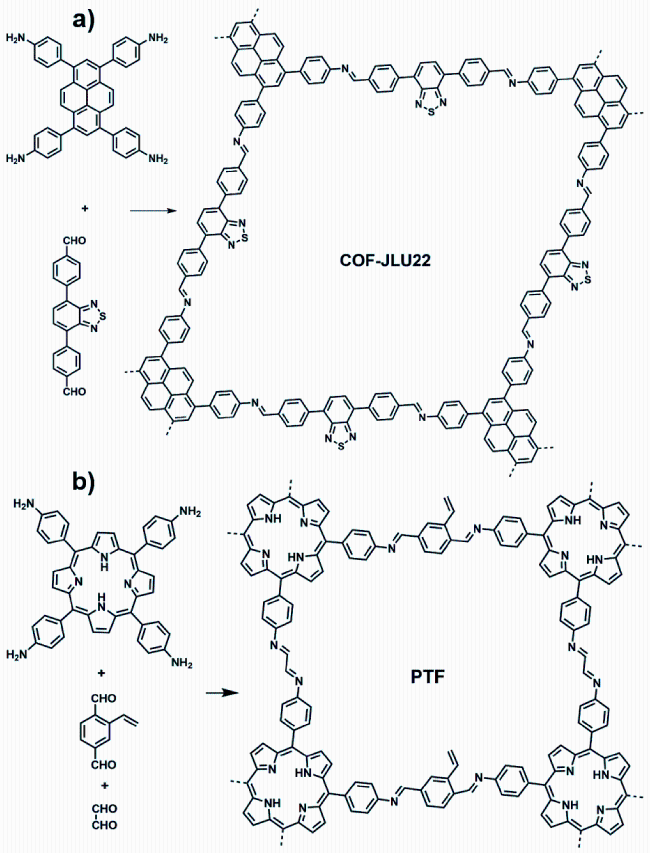
4.2.2 硝基还原
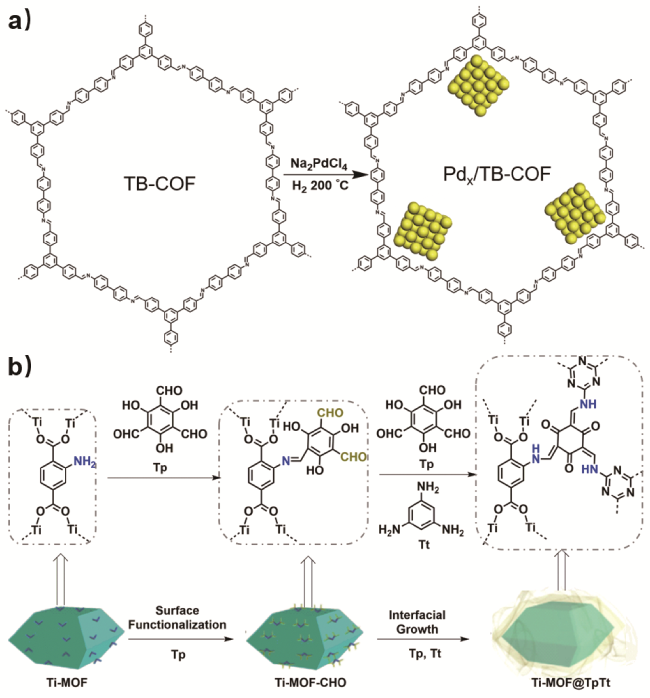
4.2.3 烯烃还原
4.3 偶联反应
4.3.1 交叉脱氢偶联反应
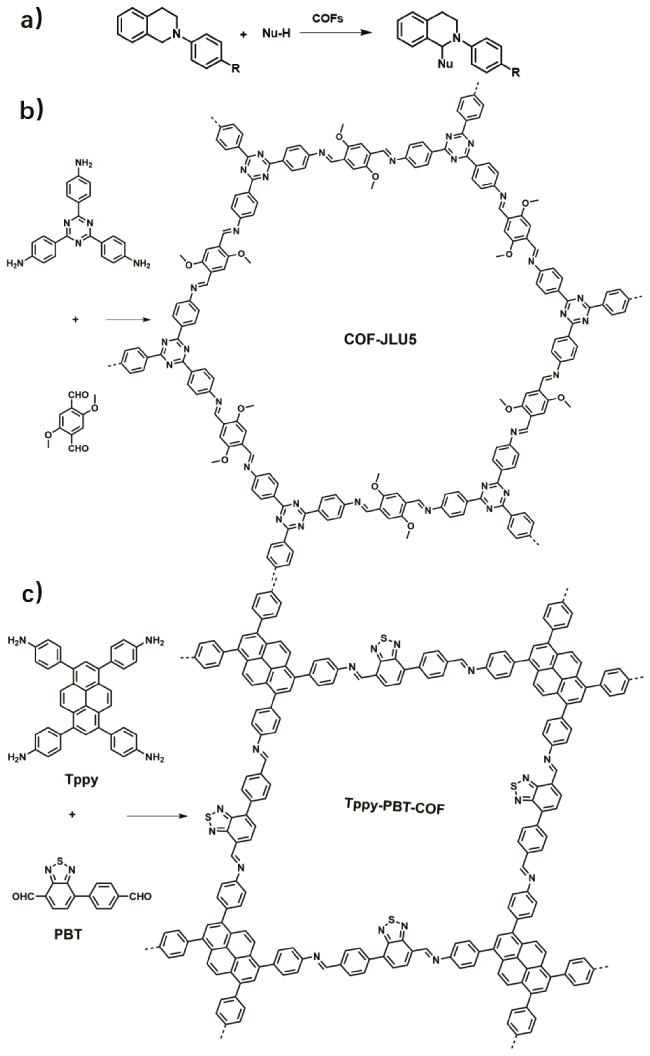
4.3.2 碳氮偶联反应

4.3.3 碳硫偶联反应
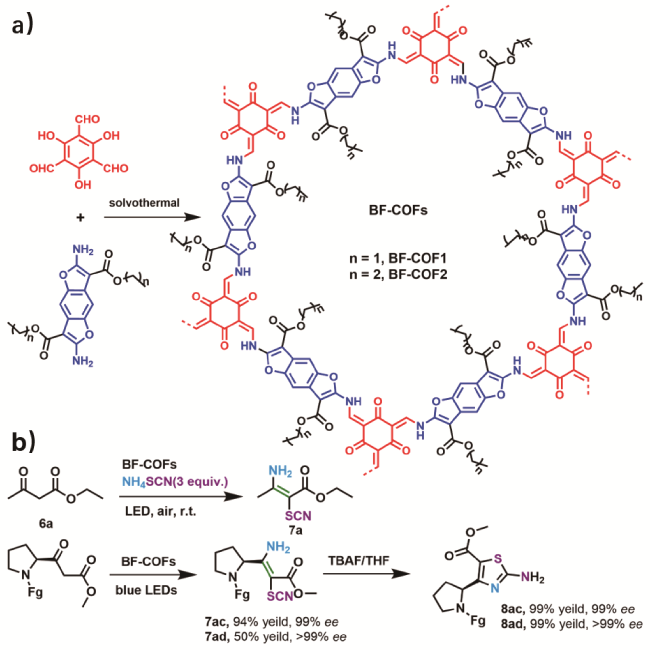
4.4 环化反应

图20 a)通过2D-COF-1光催化氧化N-S环化反应[105];b)COFs光催化环化反应机理[105];c)Py-BSZ-COF的电子结构[106]Fig. 20 a) Oxidative construction of an N-S bond by 2D-COF-1[105]. Copyright 2019, Wiley-VCH. b) Possible mechanism for sunlight-promoted aerobic oxidative construction of N-S bond[105]. Copyright 2019, Wiley-VCH. c) Electronic structure of Py-BSZ-COF[106]. Copyright 2020, American Chemical Society |

图21 a)BTT-TPA-COF的合成;b)H2P-Bph-COF光催化环加成反应[108];c)Por-Ad-COF光催化化[3+2]环加成反应[109]Fig. 21 a) Synthesis of BTT-TPA-COF. b) H2P-Bph-COF promoted photocatalytic aerobic annulation reaction for tetrahydroquinolines synthesis[108]. Copyright 2022, Elsevier. c) Por-Ad-COF promoted photocatalytic dipolar [3+2] cycloaddition reaction for pyrrolo[2,1-a]isoquinoline synthesis[109]. Copyright 2023, Wiley-VCH |
4.5 聚合反应
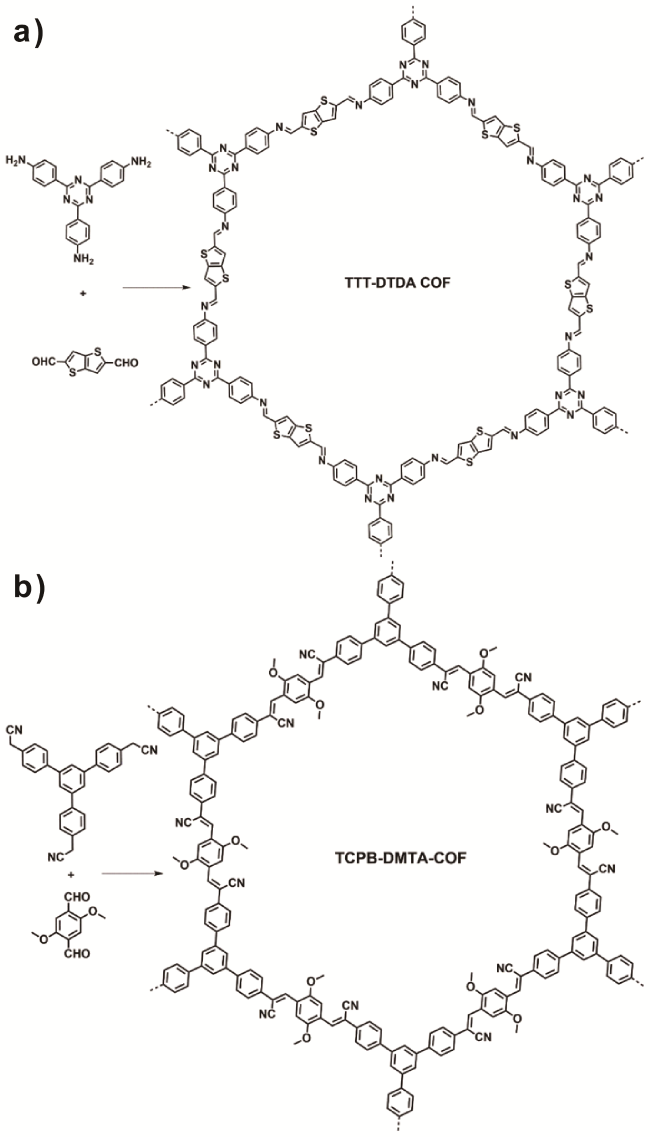
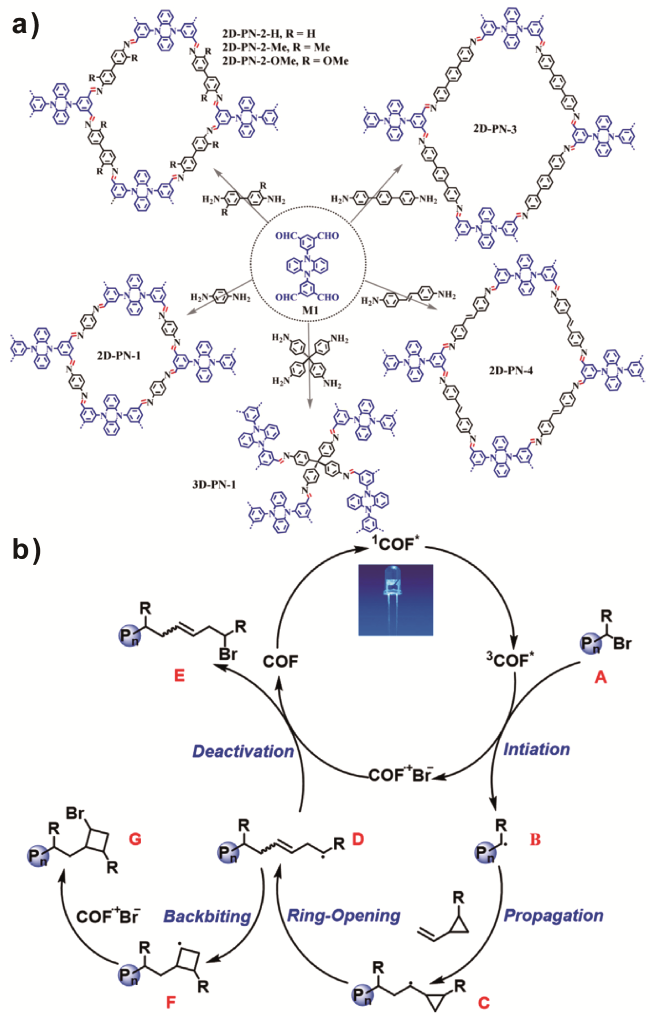
4.6 不对称光催化反应
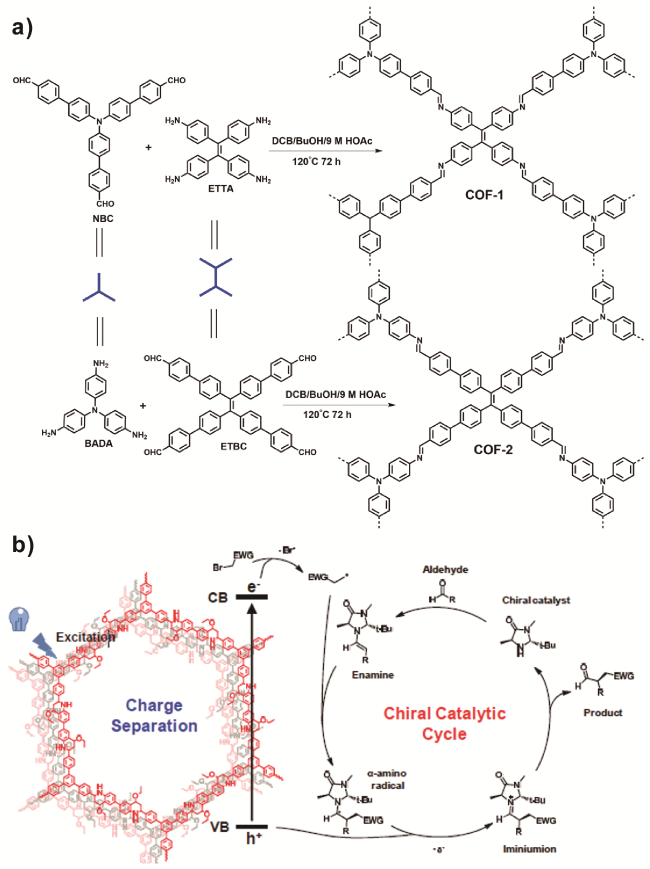
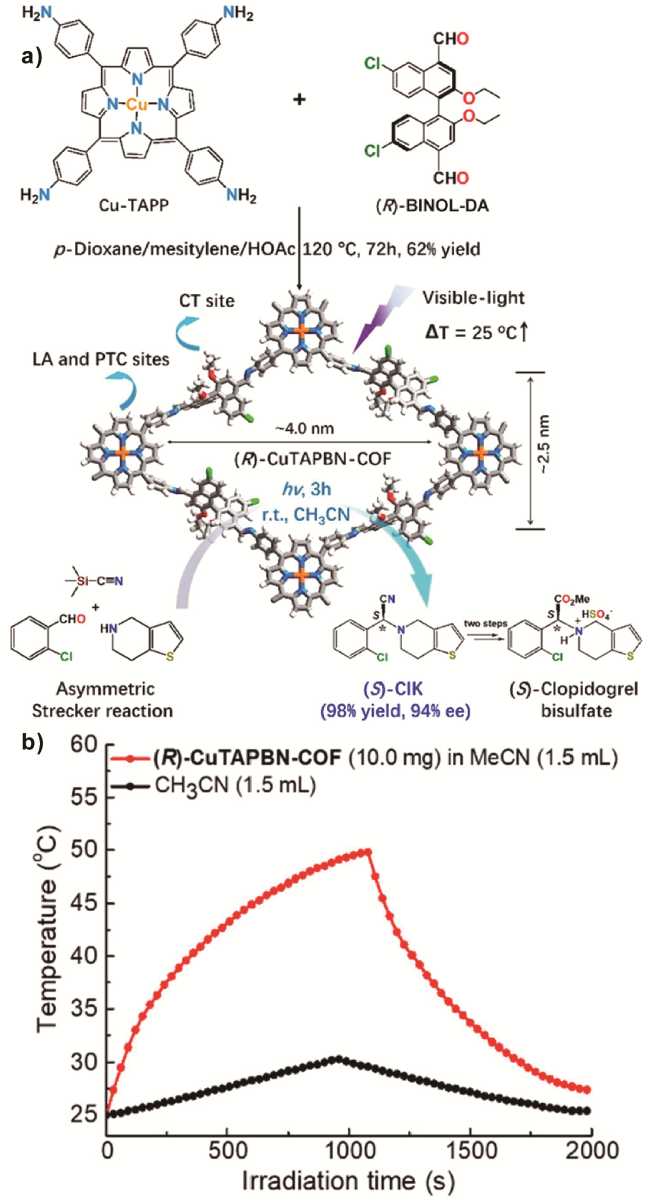
图25 a)(R)-CuTAPBN-COF合成及晶体结构和催化合成(S)-CIK的图解[120];b)(R)-CuTAPBN-COF在乙腈中的光热行为[120]Fig. 25 a) Synthesis and crystal structure of (R)-CuTAPBN-COF, and diagram representation of the catalytic synthesis of (S)-CIK[120]. Copyright 2020, American Chemical Society. b) Photothermal behavior of (R)-CuTAPBN-COF in CH3CN[120]. Copyright 2020, American Chemical Society |
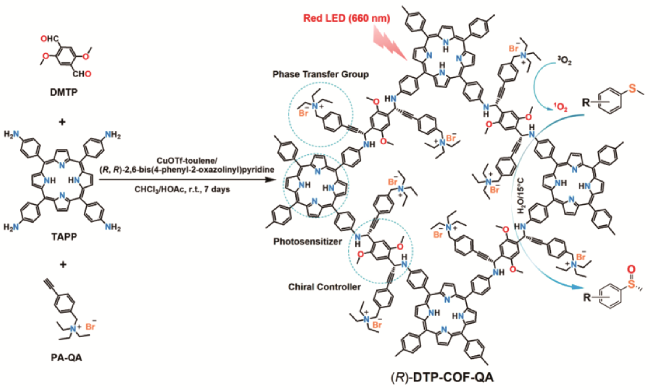
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
5 结论与展望
 唑、三聚茚酮、芘、苯并呋喃、苯并噻吩、三苯胺、四苯乙烯、苯基吩嗪、噻吩并噻吩等具有优异的吸光性能的结构,这能有效地增强COFs对可见光的吸收和光活性。在结构设计上利用电子给体-受体的结构来控制COFs的光电带隙结构,促进光生电荷的分离和传输。对于COFs的连接方式,选择共轭程度高的化学键,如亚胺键、碳碳双键、苯并
唑、三聚茚酮、芘、苯并呋喃、苯并噻吩、三苯胺、四苯乙烯、苯基吩嗪、噻吩并噻吩等具有优异的吸光性能的结构,这能有效地增强COFs对可见光的吸收和光活性。在结构设计上利用电子给体-受体的结构来控制COFs的光电带隙结构,促进光生电荷的分离和传输。对于COFs的连接方式,选择共轭程度高的化学键,如亚胺键、碳碳双键、苯并  唑等,能保证形成长程有序π共轭结构,实现光子和电子的有效传导。
唑等,能保证形成长程有序π共轭结构,实现光子和电子的有效传导。