



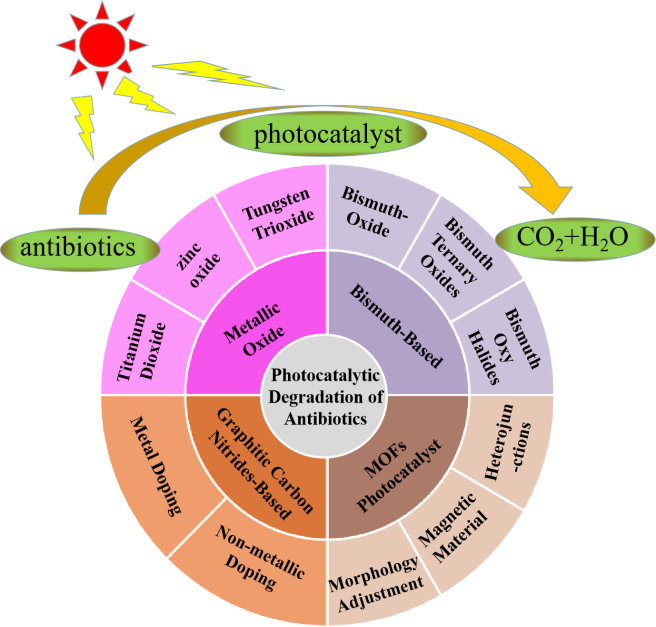
1 引言
抗生素污染物对水资源的威胁被认为是当今世界范围内最严重的环境问题之一。近年来,由于新冠病毒感染大流行,全球抗生素的使用急剧增加[1]。抗生素污染不仅会对环境产生破坏,对人类健康也有很大的潜在危害。抗生素污染物易溶于水,具有很强的流动性。传统水污染处理方法中,沉淀、絮凝、混凝和过滤等物理方法对抗生素不能起到破坏作用,如果不经其他方法处理,会造成二次污染[2];超滤、反渗透[3]和活性炭吸附[3,4]等技术主要问题在于存在膜污染、成本高、稳定性低和回收能力差等缺点,同样也不能从根本上完全破坏抗生素,达到完全治理的目标。近年来,光催化降解抗生素技术因其低成本、高效率和环境友好性,引起了研究者的广泛关注。大多数抗生素由于其分子结构稳定而很难被单一光催化材料分解。因此,通过对单一光催化材料进行改性,减小带隙宽度,尽可能吸收更多光,通过与不同能级和带隙能量的半导体形成异质结结构,抑制载流子的重组,以获得更高的光降解效率,成为目前光催化剂的研究热点。另外,近年来利用抗生素废水制氢,同时实现抗生素废水降解的研究工作也比较多[5⇓⇓⇓⇓⇓⇓~12]。
2 废水中抗生素对环境和人类健康的影响
抗生素是天然的、合成的或半合成的一种化合物,可以抑制微生物的生长或代谢活动,这些化合物是具有抗菌、抗真菌和抗寄生虫作用的生物活性分子,常用于治疗人和动物的细菌感染,也用作饲料添加剂或兽医疾病预防[15]。常见抗生素主要有四环素(TC)、环丙沙星(CIP)、磺胺甲唑(SMZ)、甲硝唑(MTZ)和头孢甲氧霉素(CFX)等。
在一些国家,抗生素不仅用于动物治疗,还用于加速植物生长和提高其产量,食物在动物体内不能完全消化,从而抗生素可能会从动物粪便中释放出来,然后这些粪便可能会被用作农业中的肥料或倾倒到废水中,进一步对人类造成伤害(图1 )。

3 光催化技术降解抗生素原理
光催化是一种低成本的有机物净化绿色技术(图2 )。光催化过程需要由作为光催化剂的半导体(Semi)吸收特定波长的光,导致电子(e−)从光催化剂的价带(VB)激发到导带(CB),在VB中产生正空穴(h+),如式(1)所示。VB中的空穴分裂H2O分子,产生羟基自由基如式(2)所示。而CB中的电子(e−)被氧分子捕获,形成超氧自由基如式(3)所示。
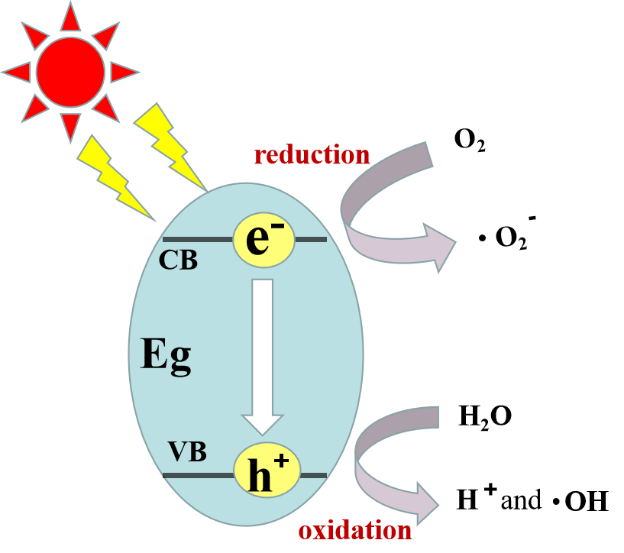
$\mathrm{Semi}+\mathrm{hv}\to \mathrm{Semi}(\mathrm{h})_{\mathrm{VB}}^{+}+\mathrm{e}_{\mathrm{CB}}^{-}$
$\mathrm{Semi}(\mathrm{h})_{\mathrm{VB}}^{+}+{{\mathrm{H}}_{\mathrm{2}}}\mathrm{O}\to \mathrm{Semi}+{{\mathrm{H}}^{+}}+\cdot \mathrm{OH}$
$\mathrm{e}_{\mathrm{CB}}^{-}+{{\mathrm{O}}_{2}}\to \cdot \mathrm{O}_{\mathrm{2}}^{-}$
$\cdot \mathrm{OH}+\mathrm{pharmaceuticals}\to \mathrm{C}{{\mathrm{O}}_{2}}+{{\mathrm{H}}_{\mathrm{2}}}\mathrm{O}$
4 常用的抗生素降解光催化材料
4.1 金属氧化物基光催化剂
TiO2是最常用于光催化的金属氧化物,常用的晶体结构为锐钛矿型,它相较于具有4个Ti—O键的金红石相而言,其八面体畸变比较明显,锐钛矿具有两个Ti—O键使得锐钛矿更容易形成缺陷,产生更多的电子空穴,有利于光催化的反应过程。但TiO2的载流子复合率较高,使用改性剂可有效提高TiO2光催化活性,改性剂可以是阳离子型或阴离子型。改性剂的引入会影响锐钛矿相的稳定性、粒径等,导致带隙缩小并吸收光谱中可见范围内的光。
金属离子可以通过调节复合速率来影响光催化剂的光活性。添加Au后的TiO2纳米管,与纯TiO2纳米管相比有更大的比表面积,Au-TiO2光催化活性得到了增强,其增强机制主要为:在Vis照射下,Au纳米颗粒(NPs)的局域表面等离子体共振(LSRP),产生光激发电子和空穴,然后高能电子注入到TiO2的导带中引发光催化反应,在Au-TiO2纳米管阵列上使用纳米线对林可霉素进行光催化降解,在Vis照射下20 min可降解83%[22]。Du等[23]也发现使用Ag掺杂的TiO2纳米管,在Vis照射60 min内,CIF的降解率达到78.4%。Li等[24]制备了金和银离子共掺杂的TiO2纳米棒,发现棒状结构的TiO2不仅能很好地与醋酸纤维素(CA)体结合,还能与CA膜保持良好的结晶度,提高孔隙率。在Vis照射下,TC的降解在120 min内达到80%。
与金属离子相比,非金属不易形成复合中心,因此它们能更有效地增强光催化活性。这种效应在多组分阴离子掺杂中尤其明显。例如,C-N-S三掺杂TiO2材料具有较大的比表面积,晶格中的一些Ti原子被C取代,使Ti-O-Ti的网络结构变为Ti-O-C,但C原子不能取代TiO2晶格中的O原子,S6+取代晶格中的Ti4+比用S2−取代O2−更容易,这种取代也有助于通过引入带间间隙和降低导带来降低TiO2的带隙能量,N被置换到O的位置,价带能级增大,而TiO2的带隙能量可以随着氮掺杂而降低。并且碳、氮和硫被添加到TiO2催化剂中,它们的电子结构发生了变化。在Vis范围内对TC分解表现出良好的光催化活性(96%,120 min内Vis照射)。并且所得材料也可以很好地对其他抗生素进行降解,如CIF(94%,150 minVis照射)和氯霉素(60%,150 minVis照射)[25]。
ZnO是另一种半导体材料,六角纤锌矿结构是ZnO最常见的结构,它是由一个锌原子与四个氧原子组成四面体结构,以此为基础构成纤锌矿,具有更高的纤盐矿形式结晶倾向,有两个晶格参数a(0.3296 nm)和c(0.52065 nm),其中Zn2+和O2−沿c轴交替排列,形成六边形亚晶格图案。在实际制备ZnO纳米材料时,所得ZnO总存在数目不同、各种形式的晶体缺陷,如氧空位(Vo)、锌空位(VZn)、氧代Zn位缺陷(OZn)等[26]。ZnO特别是在中性pH下用于光催化抗生素降解时,具有很好的量子效率和较高的光催化效率。但是,光生电子-空穴对的高复合率限制了ZnO在光催化中的应用。研究表明,在ZnO中掺杂Ag等金属或N和C等非金属可增强光催化抗生素降解的活性[27]。半导体耦合是通过与不同能级和带隙能量的半导体形成异质结,由于它们不同的带结构和能量,耦合光催化剂中光诱导电子的转移,可以实现有效的电荷分离,延长了光生成载流子的寿命[28]。宽带隙的ZnO的吸收光阈值为390 nm 的紫外光,可以通过负载稀土氧化物,通过两种稀土氧化物之间形成了异质结以及ZnO和CeO2纳米晶体之间的相互作用降低带隙值,使吸收光波长增大,红移到Vis区,提高Vis的利用率,进而提高了抗生素的降解效率,并且可同时对头孢曲松和TC的混合溶液进行降解。由于纳米ZnO不易回收,有学者提出将Fe2O3与ZnO结合,证明了Fe2O3不影响ZnO介导的光降解,并可增加纳米结构的孔隙率和吸附性能,使整体降解性能提高了约20%[29]。
氧化钨(WO3)是一种n型半导体光催化剂,化学稳定、无毒并具有电子存储特性。此外,WO3半导体中含有化学和光化学稳定的光催化剂,由带隙约为2.8 eV的钙钛矿单元组成,其中O的2p轨道形成价带,而W的5d轨道形成导带。与O2的还原电位相比,WO3具有更多的正(约+0.5 eV vs NHE)导带边缘(即O2/O2‾=−0.33 V vs NHE)。它具有优异的电子和形态性质、良好的稳定性和光活性。已经观察到WO3的各种结构,如单斜、三斜、正交、四方、六方和八面体立方,最常见的是八面体结构,钨原子位于每个八面体分子的中心,氧原子位于角落。在加热处理下,部分W-O键断裂,导致氧分子离开表面晶格并产生氧空位[30]。WO3中的一个氧空位通常会产生两个电子,从而将W6+离子还原为W5+。然而,这些空位只有在WO3的表面晶格中以最佳浓度发现时才具有积极的方面。尽管有Vis响应,纯WO3仍面临两个主要缺点:载流子重组率大和CB边缘位置小。由于WO3的CB值为+0.5 eV,因此在任何光催化应用中进行还原过程都是低效的。这意味着WO3表面没有生成O2−,或者激发电子没有足够的潜力进行特定的还原过程,从而降低了它们的光催化效率。
形貌修饰的目的是通过改善孔隙率和体积比来增加光催化反应中心和比表面积。目前已报道了具有零、一、二维和三维结构的WO3纳米复合材料的各种形态,包括纳米管、纳米线、纳米棒、纳米板、方板状、海胆状、微花状和空心微球等。通过水热法、溶剂热法、脉冲激光沉积法、阳极氧化法、化学气相沉积法、溶胶-凝胶法和电沉积法制备了不同的纳米结构。制备WO3使用了不同的前驱体,如钨氯(WCl2、WCl4、WCl5和WCl6)、钨酸铵、钨酸钠,提供不同的形态。
WO3合成期间不同的退火温度和反应时间,也会形成不同的结构,退火温度越高(高达600℃),结晶度越高,有利于电子空穴对通过半导体的传输。Rong和Wang[31]报道了随着退火温度从300℃增加到550℃,WO3纳米结构的结晶度提高,保持单斜晶系结构完整性,但表面积会随着退火温度的升高而减小。
4.2 铋基光催化剂
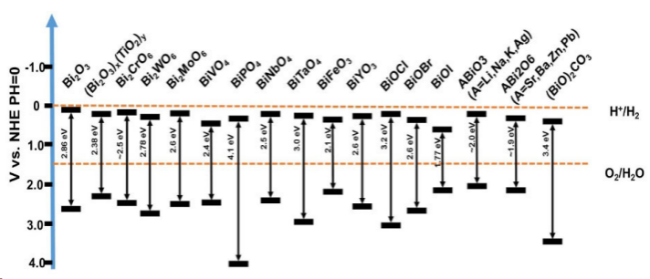
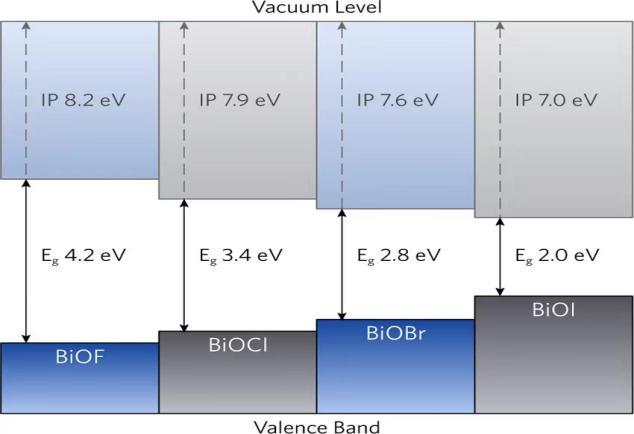
为了提高Bi氧化物的吸收率及其氧化能力,研究人员也提出了将Bi氧化物与卤化物结合,以生成氧卤化物铋(BiOX,X=F,Cl,Br,I),并获得了基本间接带隙值(图5 )[39]。但纯BiOX有时对TC、土霉素、CIF和多西环素的降解不太理想,因此常对铋基氧卤化物进行掺杂、共掺杂或偶联金属、金属氧化物、硅藻土、聚合物等[40]。Li等[41]通过简单的水解方法成功制备了BiOCl的硅藻土复合材料,硅藻土作为载体实际上相当于固体分散剂,可以将BiOCl微球均匀地固定在其表面上并防止其聚集,从而确保BiOCl的更多活性位点得以暴露,并促进光诱导载体的转移和分离。掺杂60%硅藻土所制备的BiOCl复合材料,在模拟太阳光照射下10 min内对CIP的去除效率可达到94%。
Fang等[42]设计了一种多功能集成磁性膨润土/生物/BiOBr-Bi (MB/BiOBr-Bi),通过使用支撑载体和负载Bi 的NPs来有效地去除抗生素。磁性膨润土(MB)作为光催化剂载体,具有模板样剂的功能,可将BiOBr原有的片状结构转化为分层结构。同时,表面负电荷和MB中丰富的—OH基团在[001]面暴露和氧空位生成中起着重要作用。基于这种分层结构,通过Bi还原,在BiOBr上原位均匀形成适当浓度的OVs,获得的MB/BiOBr-Bi表现出良好的光催化性能,可在80 min内有效降解92.4%的TC。
4.3 基于MOFs的光催化剂
异质结的构建对于提高MOFs的光催化效率非常有利。它不仅促进了光生电子-空穴对的分离,还增加了构成异质结的其他材料对光的吸收,以生成更多的自由电子和空穴,并提供更多的反应位点[46]。许多材料与MOFs结合以构建异质结,包括金属氧化物、金属硫化物、金属盐、无机非金属化合物、有机化合物和水滑石等。例如,Liu等[47]使用声波晶体方法制备了ZnO/ZIF-9,其中II型异质结促进了空穴-电子对的分离,与单个ZIF-9相比,TC降解率提高了两倍。在Chen等[48]的实验中,使用有机聚苯胺(PANI),通过球磨构建Z型异质结MIL-100(Fe)/PANI,MIL-100(Fe)/PANI复合材料的能带边缘也发生了轻微的红移,从单个MIL-100(Fe)的2.77 eV分别变为MP3%、MP5%、MP7%、MP9%时的2.47、2.41、2.34、2.29 eV。MIL-100(Fe)/PANI复合材料相对较窄的带隙能量可能是由于MIL-100(Fe)和PANI之间形成的杂化结构之间的强相互作用,这可以更有效地利用白光降解,通过异质结的构建,该材料对TC降解率提高了46%。此外,Abazari等[49]将水滑石应用于MOFs基异质结材料的构建,并用于SMZ的光催化降解,复合材料优异的光催化性能可能是由于Ni-Ti LDH和Zn-TMU-5的协同作用,导致对太阳光的吸收增加,光诱导载流子的有效分离,以及电荷快速转移到Ni-Ti LDH和Zn-TMU-5的反应位点和导带电位。此外,Zn-TMU-5@Ni-Ti LDH复合材料可重复使用5次,表现出了良好的可重复性。利用对苯二甲酸探针在光致发光(PL)光谱中证实了Zn-TMU-5@30% Ni-Ti LDH复合物上有OH自由基的形成。Askari等[50]制备的三元异质结CuWO4/Bi2S3/ZIF-67,对MTZ和CFX有良好降解效率(80 min的光照下为95.6%和90.1%)。
由于其多组分和可调结构,MOFs材料通常被用作模板,以此来制备出具有更好组分、形态和结构的光催化剂。同样的,Hariganesh等[52]通过煅烧MIL-101(Cr)和Cu(NO3)2的复合产物,以获得与MIL-101(Cr)相同形态的CuCr2O4/CuO,实现了良好的导电性。
长期以来,光催化剂的回收问题限制了其实际应用,许多研究人员将Fe3O4负载到光催化材料上,使其具有磁性,便于后期的回收再利用。Li等[53]生产了MIL-100(Fe)和Fe3O4的复合材料,这不仅生成了异质结结构,而且还导致复合材料内具有了磁性,Fe3O4和Fe3O4@MIL-100(Fe)表现出超顺磁特性,饱和磁化强度值分别为71.8和15.1 emu/g,并且发现8种光产物的双氯芬酸(DCF)降解过程都是从羟基化开始的。
4.4 类石墨相g-C3N4光催化剂
根据最新的研究,金属或非金属掺杂g-C3N4可以提高其光催化活性,掺杂被认为是一种有效且合适的方法[56],可以通过修饰g-C3N4的电子骨架和带隙来提高其光催化能力,因为掺杂后的光催化剂可以在价带和导带之间创建新的能级,从而提高光催化性能,这些金属或非金属作为掺杂剂有助于电子聚集,并降低e−和h+复合[57]。Wang等[58]通过双锅水热技术合成了多种新型复合材料(掺杂Cu的g-C3N4),通过光催化降解和吸附的协同作用从废水中去除TC。与纯g-C3N4光催化剂相比,Cu/g-C3N4的复合材料具有更好的吸附和光催化活性。在Vis照射下,合成的催化剂在吸附和光催化作用的协同作用下,30 min内对50 mg/LTC的降解率接近99%。此外,光催化过程中最有效的反应基团是氧自由基和氢自由基。经过5次循环后,Cu/g-C3N4表现出极大的稳定性和可重复性。铜离子与氨基之间的静电相互作用、氢键和表面络合作用是提高吸附能力的主要原因。研究还发现,掺杂可以扩大吸收波长的范围,而纯g-C3N4只能吸收蓝光。g-C3N4是一种优良的金属离子分散底物,不会因掺杂金属而产生明显的团聚现象,丰富的胺基为金属纳米颗粒的结合提供了更好的相容性,减小了金属纳米颗粒的面积,提高了稳定性。就成本而言,掺杂非金属是一种可行且有效的方法,可以提高g-C3N4的Vis吸收能力,载流子的灵活性,并简化光生e−和h+对的分离,同时保持无金属的结构[59]。Viet等[60]研究了可高效分解土霉素的Ag掺杂g-C3N4的制备,实际上,在g-C3N4上掺杂Ag后,禁带宽度从2.71 eV减小到2.46 eV,通过延迟光诱导e−和h+偶联的复合,提高了光催化活性。此外,研究人员发现[61],掺杂贵金属离子优化了光催化性能,这是由于贵金属离子具有优异的电子捕获能力,光产生的电子和空穴的分离更高。
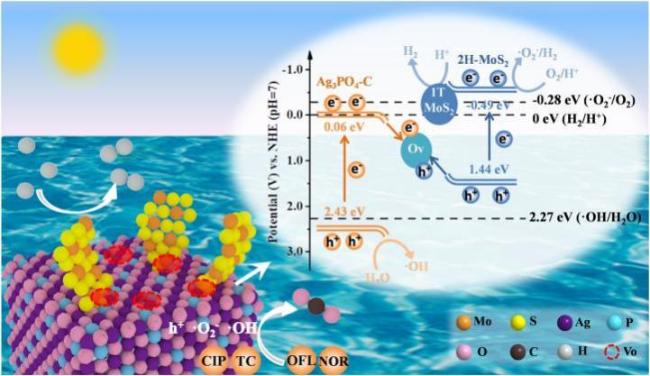
Zhang等[62]制备了Z型结构的Bi2WO6/g-C3N4复合材料并将其负载在活性碳纤维膜(ACF)上以降解金霉素,降解率可达到90.2%。这两种材料复合后,Bi2WO6的CB上被光激发的光生电子e−转移到g-C3N4的VB并与g-C3N4的VB上的空穴h+结合,延长了光生载流子的存在时间。并且g-C3N4具有相对小的CB,可以更有效地降低O2吸附在表面,并产生更多的超氧化物自由基·O2-。另外有研究人员采用微波水热法合成了一种新型层状结构的S型异质结g-C3N4/Mn(VO3)2复合光催化剂,结果表明掺杂比为1/2.75-g-C3N4/Mn(VO3)2光催化剂在SMX的降解中表现出最佳的光催化能力和优异的稳定性,在110 min时降解效率达到87.3%,分别是g-C3N4、Mn(VO3)2的11倍和14倍[63]。Zhang等[64]通过原位合成法制备了新型Ag3PO4/g-C3N4/MoSe2三元复合材料。与单一的Ag3PO4的相比,Ag3PO4/g-C3N4/MoSe2三元光催化剂对CIP和TC表现出了优异的可见光响应降解性能。
5 抗生素的光催化降解
5.1 TC的光催化降解
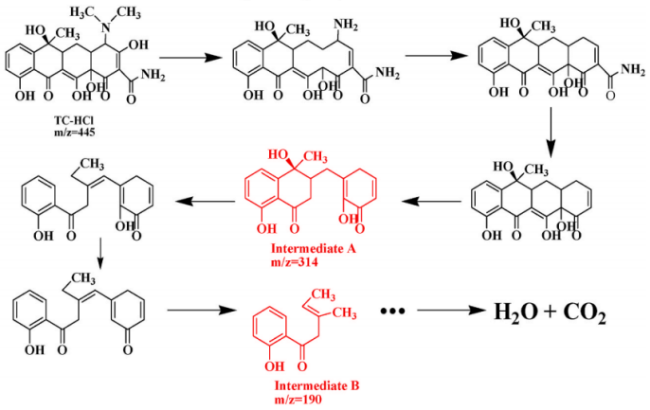
在过去的几年中,已经报道了许多通过使用不同的光催化材料来去除或降解TC的研究[69]。例如,Chen等[70]通过原位沉淀法合成了一种新型的异质结构光催化剂AgI/BiVO4,在可见光照射下对TC的分解表现出优异的光活性,TC分子在60 min内被明显消除(94.91%),在相同的实验条件下,降解效率显著优于单独的BiVO4(62.68%)和AgI(75.43%)。Wang等[71]提出了一种新型的TiO2/g-C3N4核壳量子异质结的制备方法,该方法采用了在锐钛矿型二氧化钛纳米片表面聚合量子诱变石墨氮化碳(g-C3N4)的可行策略。将其用作TC降解光催化剂,该催化剂表现出的最高TC降解速率为2.2 mg/min,其比TiO2/g-C3N4混合物高36%,比TiO2高2倍,比本体g-C3N4高2.3倍。Wang等[5]利用氯化铵在热缩合过程中分解的气泡模板效应,成功制备了具有大表面积和介孔结构的3D聚合氮化碳泡沫(CNF)。其对TC-HCl的降解在天然海水中的去除率最高,为78.9%,其次是水库水(75.0%)、自来水(62.3%)、去离子水(49.8%)、反渗透浓缩液(32.7%),然后是制药废水(18.9%)。Chen等[7]通过尿素和喹唑啉-2,4二胺(DQ)的共聚反应成功合成了芳环封端的g-C3N4纳米片。ARCNS-3可显著提高可见光驱动的光催化析氢能力,同时净化废水(1021 μmol·h−1·g−1,TC降解率为100%),远高于CNS(325 μmol·h−1·g−1,TC降解率为48%)。此外,四环素对ARCNS-3的矿化率达到92.1%,远优于CNS(18%)。Jiang等[72]设计合成了一种先进而稳定的可见光驱动(Bi)BiOBr/rGO光催化剂用于降解TC,在20 min内达到>98%的去除率。通过50 h的连续光催化操作证实了光催化剂的稳定性。在连续流动配置中,几乎100%的TC去除率可以保持约10 h(图8 )。Liu等[56]通过在g-C3N4的结构中同步引入硫和硒原子,成功地合成了硫和硒共掺杂的石墨氮化碳(SSCN)。由于引入硫和硒,SSCN的不对称结构不仅保持了π-π*电子跃迁,而且也引发了g-C3N4中的n-π*电子跃迁。SSCN-50表现出最佳的光催化性能,对抗生素(TC)和有机染料(RhB)的降解率分别为78.0%和99.4%,分别是g-C3N4的2.5倍和16.8倍。Li等[12]成功制备了具有独特纳米异质结构、能带结构和化学键界面的中空花状微球结构S型异质结In2Se3/Ag3PO4,它降解TC的光催化活性显著提高并可同时产生氢气,这种性能的提高主要归功于中空结构和S型异质结。
5.2 CIP的光催化降解
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
最后,我们比较了上面讨论的不同光催化剂,列于表1 中。大多数催化剂可以在2 h内轻松有效地去除抗生素污染物,具有相对较高的降解效率,这充分证明了光降解快速有效去除抗生素的优越性。
表1 不同催化剂降解抗生素及其降解效果Table 1 Degradation of antibiotics with different catalysts and their degradation effects |
| Antibiotic | Photocatalyst | Result | Degradation mechanism | ref |
|---|---|---|---|---|
| TC | Heterogeneous TiO2/g-C3N4 | 100 mg TiO2/g-C3N4 can decompose 20 mg tetracycline (2.2 mg/min) within 9 min | Co action of ·O2− and h+ | 71 |
| TC | Heterogeneous AgI/BiVO4 | The degradation rate of TC within 70 min is 94.91% | Co action of ·OH,·O2− and h+ | 70 |
| TC | 3D polymerized carbon nitride foam | TC degradation rate in 70 min seawater 78.9% | Co action of ·O2− and h+ | 5 |
| TC | ARCNS-3 | TC degradation rate within 5 h is 100% | Inhibition of photogenerated electrons and free radical intermediates by aromatic rings | 7 |
| TC | S-scheme In2Se3/Ag3PO4 | 1 h TC degradation rate 93.1% | Co action of ·OH,·O2− and h+ | 12 |
| TC | (Bi)BiOBr/rGO | 20 min degradation rate 98% | Co action of ·O2− and h+ | 43 |
| CIP | CuO:Zn | The degradation rate of CIP within 240 min is 94.6% | Synergistic effect of ·OH and h+ | 76 |
| CIP | Flaky peeling g-C3N4 | The degradation rate of CIP within 60 min is 78% | Co action of ·O2− and h+ | 77 |
| CIP | CZSNO20 | The degradation rate of CIP in 60 minutes is nearly 83% | Co action of e−, h+,·OH and ·O2− | 8 |
| CIP | Z-scheme Ag3PO4 @MoS2 | The degradation rate of CIP within 2 h reached 91.7% | Co action of ·OH,·O2− and h+ | 6 |
6 结论与展望
抗生素的普遍使用和过量使用不仅会对环境产生破坏,对人类健康也会有很大的潜在危害。光催化降解抗生素具有成本低、效率高、绿色环保的优点,其催化降解抗生素的机理主要取决于自由基和活性氧的形成。详细介绍了半导体金属氧化物光催化剂、铋基光催化剂、基于MOFs的光催化剂和类石墨相g-C3N4光催化剂等4种常见的光催化剂及其改性的最近新研究成果。对光催化剂的改性研究主要集中在:(1)提高光催化剂在水溶液中的分散性,增大其表面吸附、解吸药物的能力;(2)构建异质结的方法使光催化剂具有合适的带隙,拓宽其光吸收范围,抑制电子-空穴对的复合,提高光催化效果;(3)实现光催化剂从水中回收再利用,提高其利用率,降低二次污染。
然而,随着抗生素的普遍应用,抗生素种类越来越丰富,不同类型的抗生素具有不同的结构特征。由于光催化剂的VB不同,在光催化过程中与光生空穴是否可以直接氧化,通过对自由基的捕获实验等进一步探究。另外,在光降解过程中有许多中间体的产生,对其进行结构、毒性大小分析,并且通过测定其总有机碳(TOC)含量来判断矿化程度,了解光催化降解的详细过程,进一步探究其降解机理,对提高抗生素降解效率也至关重要。