




1 引言
2 抗冻蛋白的功能特性
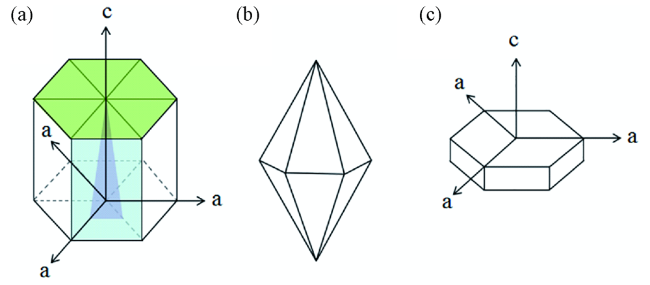
图1 冰晶示意图。(a) 冰晶的不同晶面示意图,绿色区域表示基面,蓝色区域表示主棱面,紫色区域表示锥面。(b) 冰晶在a轴方向的生长受到抑制时,形成双棱锥形状。(c) 冰晶在a轴和c轴方向的生长均受到抑制时,形成六棱盘形状Fig. 1 Schematic representation of ice crystal.(a) Different ice planes of ice crystal, the green area represents the basal plane, the blue area represents the primary prism plane and the purple area represents the pyramidal plane.(b) Ice crystal forms a hexagonal bipyramid shape, when the growth of it in the a-axis direction is inhibited.(c) Ice crystal forms a hexagonal plate shape, when the growth of it in both a-axis and c-axis directions is inhibited |
3 抗冻蛋白的结构和作用机制
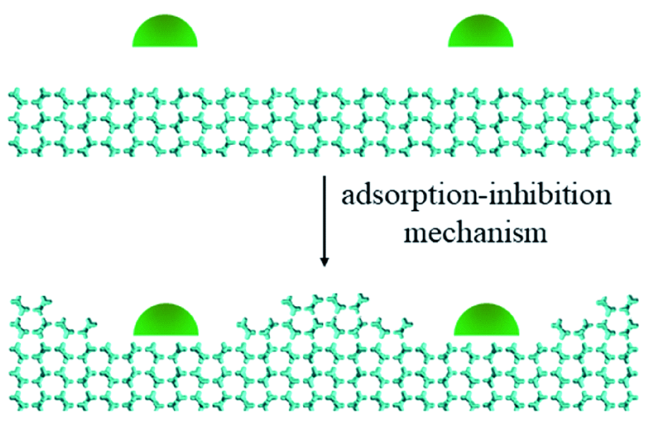
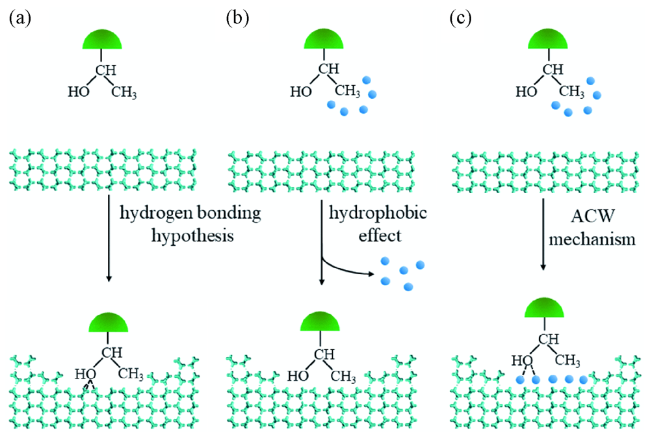
图4 抗冻机理示意图。(a) 氢键假说;(b) 疏水作用;(c) ACW机制。冰结合位点以苏氨酸为例,绿色半球体代表抗冻蛋白,青色柱体代表冰晶,蓝色小球代表锚定结合水,黑色虚线代表氢键,自由的水分子没有显示Fig. 4 Schematic representation of antifreeze mechanism.(a) hydrogen bonding hypothesis,(b) hydrophobic effect,(c) ACW mechanism. An example of ice binding site is threonine, the green hemispheres represent antifreeze proteins, the cyan pillars represent ice crystals, the blue spheres represent anchored clathrate water, the black dotted lines represent hydrogen bonds. Free water molecules are not shown |
3.1 鱼类AFPs
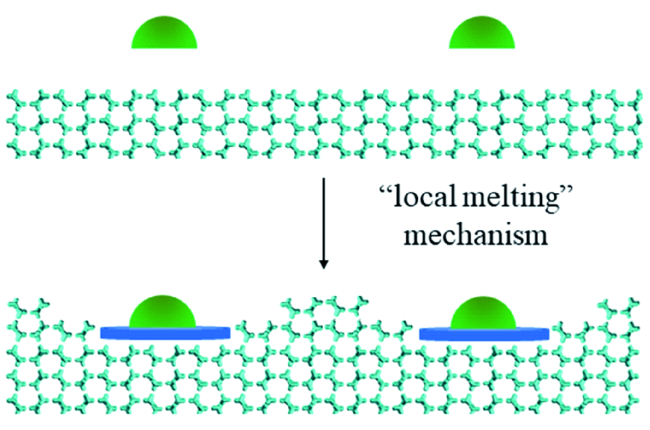
图5 局部融化机制示意图。绿色半球体代表抗冻蛋白,青色柱体代表冰晶,深蓝色区域代表过冷水,自由的水分子没有显示Fig. 5 Schematic representation of “local melting” mechanism, the green hemispheres represent antifreeze proteins, the cyan pillars represent ice crystals, the deep blue area represents supercooled water. Free water molecules are not shown |
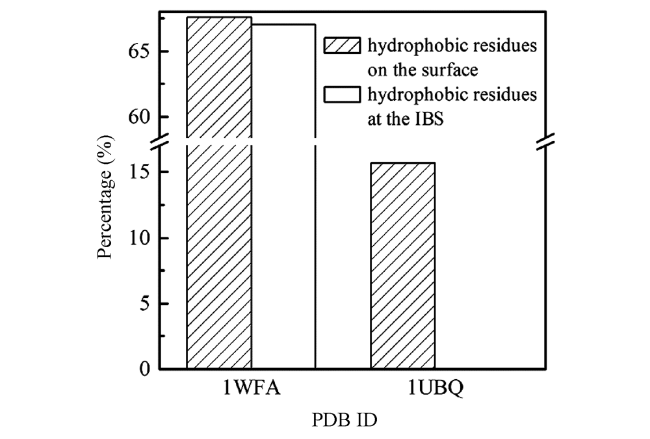
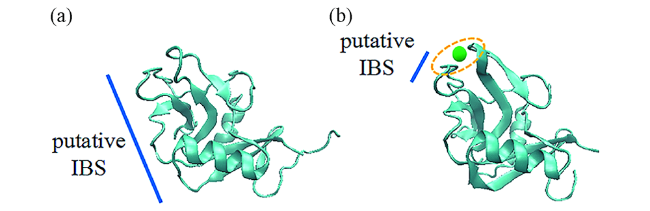
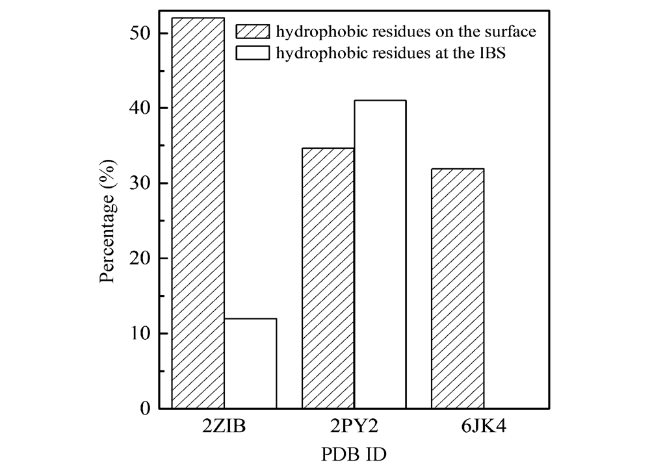
图7 (a) Ca2+-非依赖型BrAFP (PDB ID: 2ZIB)和(b) Ca2+-依赖型抗冻蛋白hAFP (PDB ID: 2PY2)的假定冰结合平面对比图,绿色的小球代表Ca2+,黄色虚线区域代表Ca2+结合环Fig. 7 Comparison of putative IBSs of(a) Ca2+-independent BrAFP(PDB ID: 2ZIB) and(b) Ca2+-dependent hAFP(PDB ID: 2PY2), the green sphere represents Ca2+ and the yellow dotted line area represents Ca2+-binding loop |
3.2 昆虫AFPs
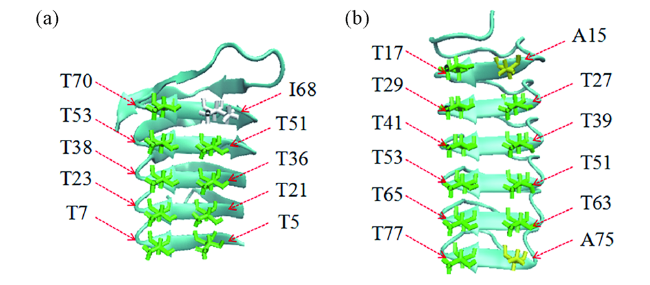
图10 2种TxT基序的昆虫抗冻蛋白(PDB ID: 1L0S,1EZG)冰结合序列示意图Fig. 10 Ice-binding sites of two insect AFPs with TxT motifs(PDB ID: 1L0S,1EZG) |
表1 抗冻蛋白的结构特征、性质和机理总结Table 1 Summary of structural characteristics, properties and mechanisms of antifreeze proteinsa |
| Origin and type | Structural characteristics | PDB ID | R1 (%)b | R2 (%)C | R3 (%)d | R4 (%)e | Antifreeze Mechnism | ref. | |
|---|---|---|---|---|---|---|---|---|---|
| Fish | AFPⅠ | α-helix, 65% alanine | 1WFA | 68 | 33 | 14 | 0 | a hydration mediated AFP adsorption mechanism/“local melting” mechanism | 33, 79 |
| AFPⅡ | Globular, 2α-helix+2 β-sheet | 2ZIB | 52 | 12 | 24 | 38 | ACW mechanism | 84, 88 | |
| 2PY2 | 35 | 41 | 28 | 24 | |||||
| 6JK4 | 32 | 0 | 26 | 50 | |||||
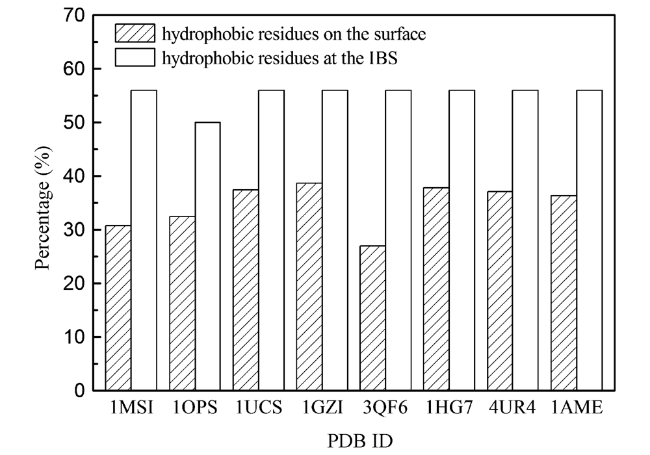
| AFPⅢ | Globular, no dominant nonpolar amino acids | 1MSI | 31 | 56 | 19 | 0 | a hydration mediated AFP adsorption mechanism | 67, 92, 93, 95 | |
| 1OPS | 32 | 50 | 14 | 0 | |||||
| 1UCS | 38 | 56 | 19 | 0 | |||||
| 1GZI | 39 | 56 | 19 | 0 | |||||
| 3QF6 | 27 | 56 | 19 | 0 | |||||
| 1HG7 | 38 | 56 | 16 | 0 | |||||
| 4UR4 | 37 | 56 | 15 | 0 | |||||
| 1AME | 36 | 56 | 16 | 0 | |||||
| AFPⅣ | 4 antiparallel helical bundles, Glu-rich | The Adsorption-Inhibition Hypothesis | 98 | ||||||
| AFGP | (Ala-Thr-Ala)n with a disaccharide moiety(Galβ1- 3GalNAcα1-) attached to each Thr residue | The Adsorption-Inhibition Hypothesis/Perturbation of Long- Range Water Dynamics | 17, 29, 31, 34, 35 | ||||||
| Insect | Polyproline type II | 3BOI | 34 | 47 | 18 | 7 | “local melting” mechanism | 104 | |
| 2PNE | 38 | 47 | 21 | 7 | |||||
| β-solenoid, seven coils of TCTxSxxCxxAx repeats | 1EZG | 16 | 44 | 14 | 0 | ACW mechanism | 28, 76 | ||
| β-solenoid, flat β-sheet of Thr-x-Thr motifs | 1EWW | 19 | 27 | 19 | 0 | 16, 69 | |||
| 1L0S | 21 | 20 | 19 | 0 | |||||
| 1M8N | 24 | 33 | 16 | 0 | |||||
| Flat β-solenoid | 4DT5 | 13 | 29 | 20 | 4 | ||||
| β-solenoid, Thr-rich, Cys- rich | 1L1I | 15 | 44 | 15 | 0 | ||||
| Plant Micro-organism | Bacteria | β-solenoid | 3ULT | 15 | 50 | 14 | 0 | ACW mechanism | 50 |
| β-solenoid | 3P4G | 20 | 5 | 29 | 18 | ACW mechanism | 32 | ||
| β-solenoid with a triangular cross-section alongside an α-helix | 3WP9 | 21 | 32 | 25 | 10 | ACW mechanism | 15, 118, 119 | ||
| 4NU2 | 28 | 38 | 16 | 5 | |||||
| 6EIO | 32 | 20 | 8 | 10 | |||||
| Fungus | β-solenoid with a triangular cross-section alongside an α-helix | 3UYU | 24 | 41 | 22 | 13 | ACW mechanism | 14, 51, 120, 121 | |
| 5B5H | 24 | 48 | 6 | 8 | |||||
| 6A8K | 24 | 50 | 10 | 5 | |||||
| 3VN3 | 25 | 35 | 13 | 12 | |||||
| Ubiquitin | Globular | 1UBQ | 16 | 41 | Non-antifreeze protein(as a reference) | ||||
a 29 wild-type antifreeze proteins which have crystal structure in PDB database. | |
b R1: the ratio of hydrophobic residues on the surface. The relative solvent accessibility of each residue of antifreeze proteins is calculated, and the residues with the relative solvent accessibility more than 20% are regarded as the surface residues, and the ratio of hydrophobic residues to surface residues is calculated. | |
C R2: the ratio of hydrophobic residues at the ice-binding site(IBS). | |
d R3: the ratio of charged residues on the surface. | |
e R4: the ratio of charged residues at the ice-binding site(IBS). The Ca2+-dependent AFP, 6JK4, only contains four residues at its IBS[84], two of which are negatively charged, leading to a relatively high R4. |
3.3 植物AFPs
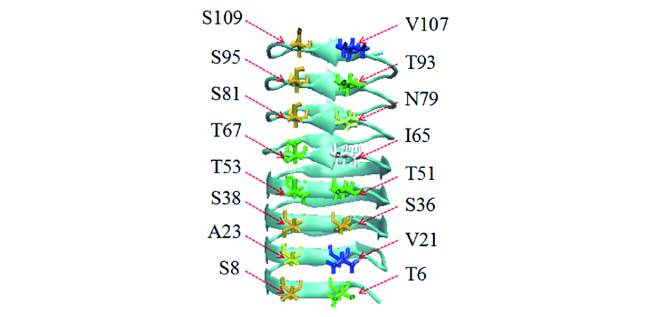
3.4 微生物AFPs
3.5 不同种类AFPs抗冻机制的异同
3.6 影响抗冻活性的结构因素
4 抗冻蛋白仿生材料

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
图12 仿生抗冻材料。(a) 不同形状的抗冻金胶体[135]。(b)氧化准碳氮化物量子点OQCNs[136]。(c) 昆虫TmAFP(上)和合成染料自组装形成的超分子冰生长抑制剂(下)的侧面(左)和正面(右)对比示意图[11],红色小球代表羟基,蓝色小球代表氮原子,灰色部分代表碳骨架。(d)基于自组装肽的超分子冰生长抑制剂示意图[137]Fig. 12 Bioinspired antifreeze materials.(a) different shapes of antifreeze gold colloids[135]. Copyright(2019) American Chemical Society.(b) oxidized quasi-carbon nitride quantum dots OQCNs[136]. Copyright John Wiley & Sons.(c) side and front views of TmAFP, an AFP from insects, and a supramolecular ice growth inhibitor[11], the blue spheres represent the amine groups, the gray part represents the carbon skeleton and the red spheres represent the hydroxyl groups. Copyright 2016, American Chemical Society.(d) schematic of the supramolecular ice growth inhibitors based on self-assembling peptides[137]. Copyright 2019, American Chemical Society |