



1 引言
2 氨硼烷的性质和合成
2.1 氨硼烷的性质
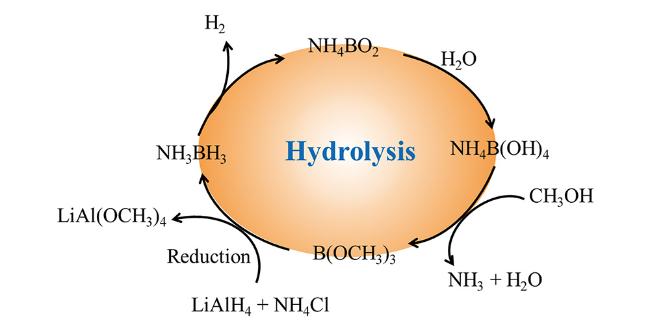
2.2 氨硼烷的合成
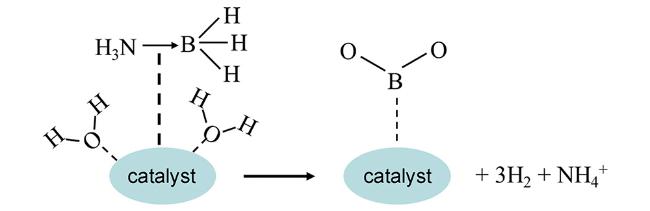
3 催化氨硼烷水解反应机理
4 氨硼烷水解制氢金属催化剂
4.1 贵金属催化剂
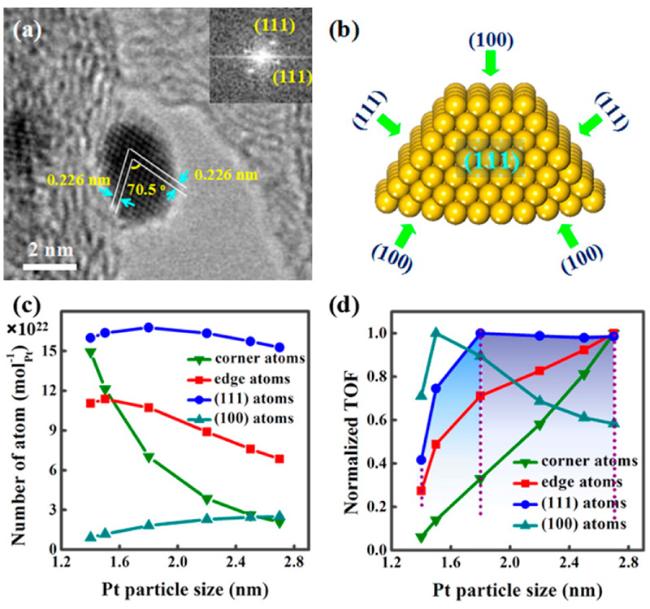
图2 (a) Pt/CNT催化剂的高分辨TEM图;(b)截断八面体的示意图;(c) Pt颗粒表面不同位置原子数量随颗粒尺寸变化的关系图;(d) 不同位置原子归一化的TOF随颗粒尺寸变化的关系图[50]Fig.2 (a) Typical HRTEM image of Pt nanoparticle supported on CNT.(b) Schematic diagram of truncated cuboctahedron.(c) Plots of number of surface atoms per mole of Pt with Pt particle size of truncated cuboctahedron.(d) Plots of normalized TOF with Pt particle size[50]. Reprinted with permission from ref [50]. Copyright 2014, American Chemical Society |
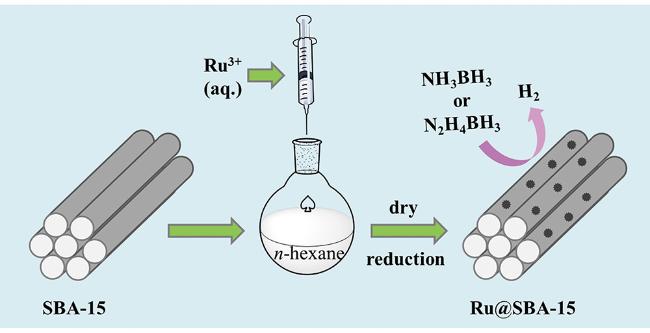
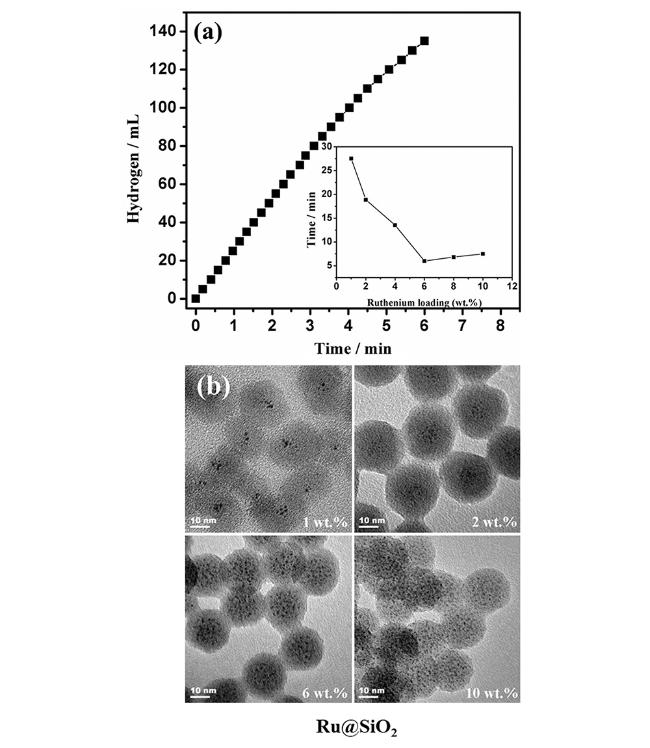
图5 Ru@SiO 2催化剂(Ru负载量:6 wt%;[Ru]:0.5 mM)催化氨硼烷水解制氢性能图;插图是不同Ru负载量与时间关系图;(b) 不同金属负载量的Ru@SiO2催化剂的TEM图[73]Fig.5 (a) Hydrogen generation from hydrolysis of AB(200 mM, 10 mL) by Ru@SiO 2 nanospheres(Ru loading=6 wt% and [Ru]=0.5 mM) at 298 K. The inset shows the reaction time versus the loading of ruthenium;(b) Representative TEM images of the Ru@SiO2 nanospheres with different Ru loadings[73]. Reprinted with permission from ref [73]. Copyright 2014, Elsevier |
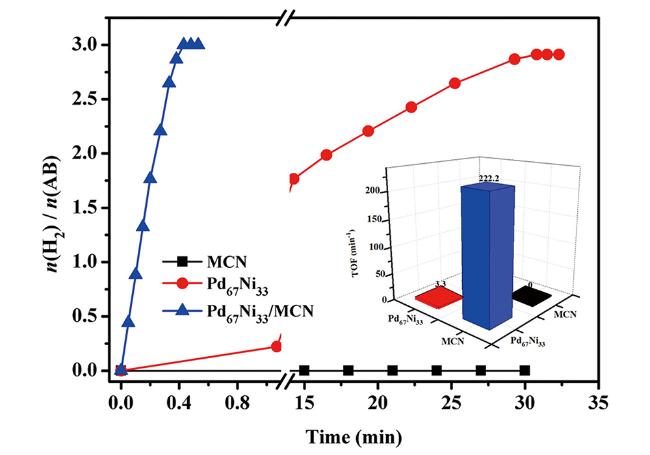
图6 MCN、Pd 67Ni 33与Pd 74Ni 26/MCN催化剂在室温条件下催化氨硼烷水解制氢性能图;插图是不同催化剂对应的TOF值图[79]Fig.6 Time plots for hydrogen release from aqueous AB solution(200 mM, 5 mL) catalyzed by MCN, Pd 67Ni 33, and Pd 74Ni 26/MCN NCs at room temperature. Inset: the corresponding TOF values of the catalysts[79]. Reprinted with permission from ref [79]. Copyright 2018, Wiley-VCH |
表1 不同贵金属催化剂催化氨硼烷水解制氢的催化性能对比Table 1 Catalytic performance of noble metal catalysts for hydrogen generation from the hydrolysis of ammonia borane |
| Catalyst | T/K | n metal/ n AB | TOF/min -1 | E a/kJ·mol -1 | ref |
|---|---|---|---|---|---|
| Commercial 20 wt% Pt/C | RT | 0.018 | 83.3 | - | 19 |
| Pt/γ-Al 2O 3 | RT | 0.018 | 222.2 | 21 | 48 |
| Pt@MIL-101 | 298 | 0.0029 | 413.8 | - | 49 |
| Pt/CNTs | 298 | - | 407.2 | 38 | 50 |
| Pt@CeO 2 nanonecklace | RT | 0.018 | 133.3 | - | 51 |
| Pt-CeO 2/rGO | 298 | 0.0079 | 93.8 | 64.7 | 52 |
| Pt 2/graphene | 300 | 0.0011 | 2800 | - | 53 |
| Pt@SiO 2 | 303 | 0.00245 | 158.6 | 53.9 | 74 |
| Pt@PC-POP | 303 | - | 104.33 | 56.42 | 85 |
| Pt25@TiO 2 | 298 | 0.0016 | 311 | - | 86 |
| Pt@h-mNSiO 2 | 298 | 0.0013 | 371.7 | 49.4 | 87 |
| Rh/γ-Al 2O 3 | RT | 0.018 | 128 | 21 | 48 |
| Rh/NaY | 298 | 0.002 | 92 | 66.9 | 59 |
| Rh/CeO 2 | 298 | 0.0008 | 2010 | 42.6 | 62 |
| Rh/AC | 298 | 0.0038 | 188 | 39.9 | 63 |
| Rh/graphene | 298 | 0.004 | 325 | 19.7 | 64 |
| Rh/CNTs | 298 | 0.0025 | 706 | 32 | 65 |
| Rh@S-1-H | 298 | 0.0011 | 699 | 75.5 | 66 |
| In situ Rh/C | 298 | 0.0114 | 1246 | 40.9 | 88 |
| Rh@UiO-66 | 298 | 0.0056 | 219.8 | 38.4 | 89 |
| Ru/γ-Al 2O 3 | RT | 0.018 | 55.5 | 23 | 48 |
| Ru/Carbon black | 298 | 0.0085 | 429.5 | 34.81 | 69 |
| Ru/C-300 | 298 | 0.0034 | 643 | 38.7 | 70 |
| Ru/Ce(OH)CO 3 | 298 | 0.0069 | 389.6 | 60.16 | 71 |
| Ru@SBA-15 | 298 | 0.0025 | 231 | 34.8 | 72 |
| Ru@SiO 2 | 298 | 0.002 | 200 | 38.2 | 73 |
| Ru/MWCNT | 298 | 0.00094 | 329 | 33 | 90 |
| Ru/graphene | 298 | 0.002 | 600 | 12.7 | 91 |
| Ru/g-C 3N 4 | 298 | 0.0013 | 384.6 | 37.4 | 92 |
| Metastable Ru | 298 | 0.0025 | 21.8 | 27.5 | 93 |
| Pd/RGO | 298 | 0.004 | 6.25 | 51 | 76 |
| Pd/MCN | 298 | 0.03 | 125 | 57 | 79 |
| Pd/SiO 2-CoFe 2O 4 | 298 | 0.0019 | 254 | 52 | 84 |
| Ag/SBA-15 | 298 | 0.093 | 12 | - | 80 |
| Ag/SiO 2-CoFe 2O 4 | 298 | 0.0028 | 264 | 53.4 | 82 |
4.2 非贵金属催化剂
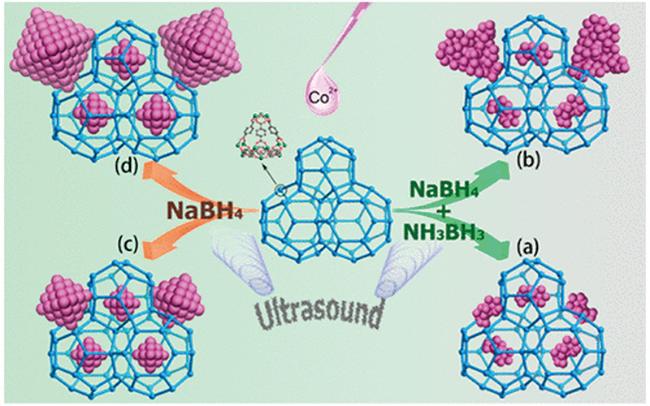
图7 (a) Co/MIL-101-1-U,(b) Co/MIL-101-1,(c) Co/MIL-101-2-U,(d) Co/MIL-101-2催化剂的合成示意图[104]Fig.7 Schematic illustration for the synthesis of four types of MIL-101-supported Co NPs: (a) Co/MIL-101-1-U;(b) Co/MIL-101-1;(c) Co/MIL-101-2-U;(d) Co/MIL-101-2[104]. Reprinted with permission from ref [104]. Copyright 2017, American Chemical Society |
表 2 不同非贵金属催化剂催化氨硼烷水解制氢的催化性能对比Table 2 Catalytic performance of non-noble metal catalysts for hydrogen generation from the hydrolysis of ammonia borane. |
| Catalyst | T/K | n metal/ n AB | TOF/min -1 | E a/kJ·mol -1 | ref |
|---|---|---|---|---|---|
| In situ Fe NPs | RT | 0.12 | 3.12 | - | 94 |
| Co/γ-Al 2O 3 | RT | 0.018 | 2.27 | 62 | 42 |
| In situ Co NPs | RT | 0.04 | 44.1 | - | 95 |
| Co/PEI-GO | 298 | 0.11 | 39.9 | 28.2 | 103 |
| Co/MIL-101-1-U | 298 | 0.02 | 51.4 | 31.3 | 104 |
| Co/NPCNW | 298 | 0.075 | 7.29 | 25.4 | 105 |
| Co@N-C-700 | 298 | 0.057 | 5.6 | 31 | 106 |
| Co/HPC | 323 | 0.11 | 2.94 | 32.8 | 107 |
| Co/CTF | 298 | 0.05 | 42.3 | 42.7 | 108 |
| Co NCs@PCC-2a | 298 | 0.07 | 90.1 | - | 109 |
| Co/rGO | 298 | 0.1 | 6.86 | 27.1 | 102 |
| Co@C-N@SiO 2-800 | 298 | - | 8.4 | 36.1 | 109 |
| G6-OH(Co 60) | 298 | 0.013 | 10 | 50.2 | 110 |
| Co-(CeO x ) 0.91 | 298 | 0.04 | 79.5 | 31.82 | 111 |
| Ni/γ-Al 2O 3 | RT | 0.018 | 2.5 | - | 42 |
| Ni/C | 298 | 0.0425 | 8.8 | 28 | 119 |
| Ni/SiO 2 | 298 | 0.0225 | 13.2 | 34 | 120 |
| Ni/ZIF-8 | RT | 0.016 | 14.2 | - | 123 |
| Ni@MSC-30 | RT | 0.016 | 30.7 | - | 124 |
| NiMo/graphene | 298 | 0.05 | 66.7 | 21.8 | 128 |
| Ni-CeO x /graphene | 298 | 0.08 | 68.2 | 28.9 | 135 |
| Ni@3D-(N)GFs | RT | 0.009 | 41.7 | - | 113 |
| Ni/CNT | 298 | - | 26.2 | 32.3 | 115 |
| Ni/PDA-CoFe 2O 4 | 298 | 0.017 | 7.6 | 50.8 | 116 |
| Ni/Ketjenblack | 298 | 0.13 | 7.5 | 66.6 | 118 |
| Cu/γ-Al 2O 3 | RT | 0.018 | 0.23 | - | 42 |
| Cu@Cu 2O | 293 | 0.15 | 0.32 | - | 43 |
| Zeolite confined Cu | 298 | 0.013 | 1.25 | 51.8 | 137 |
| Cu/CoFe 2O 4@SiO 2 | 298 | 0.0031 | 40 | - | 140 |
| Cu/RGO | 298 | 0.1 | 3.61 | 38.2 | 142 |
| Cu@SiO 2 | 298 | 0.08 | 3.24 | 36 | 143 |
| p(AMPS)-Cu | 303 | 0.069 | 0.72 | 48.8 | 144 |
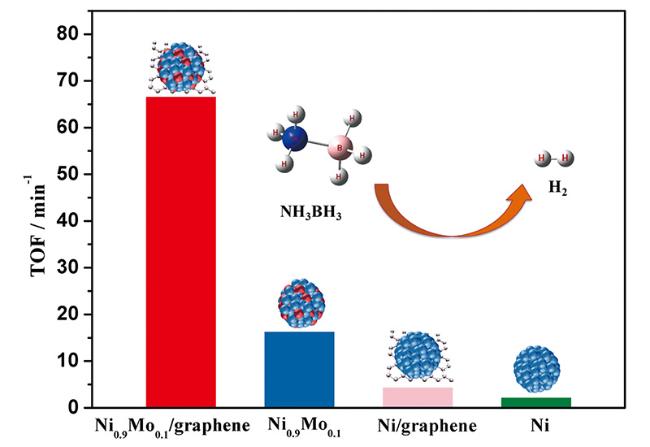
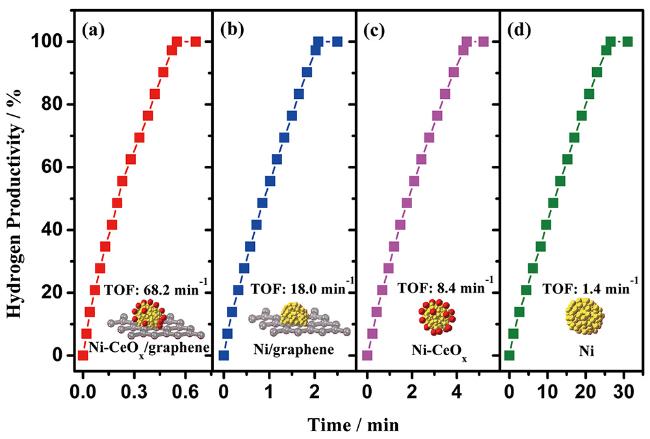
图9 不同催化剂催化氨硼烷水解制氢性能图[135]Fig.9 Hydrogen productivity vs. reaction time for hydrogen release from an aqueous AB solution(200 mM, 5 mL) catalyzed by different catalysts at 298 K( n Ni/ n AB=0.08)[135]. Reprinted with permission from ref[135]. Copyright 2018, Tsinghua University Press and Springer-Verlag GmbH Germany |
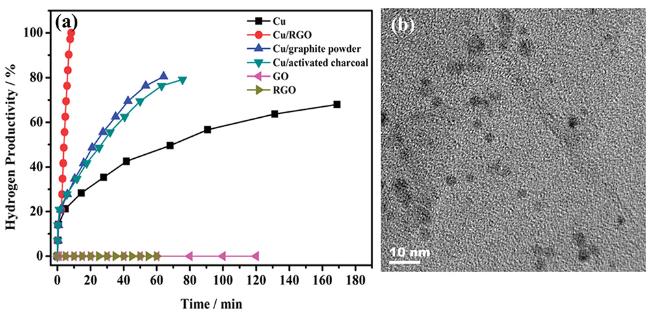
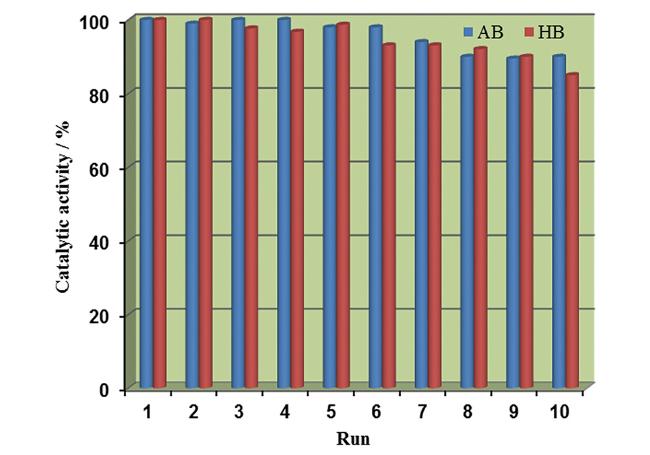
4.3 多金属协同催化剂
表 3 不同多金属协同催化剂催化氨硼烷水解制氢的催化性能对比Table 3 Catalytic performance of synergistic metal catalysts for hydrogen generation from the hydrolysis of ammonia borane |
| Catalyst | T/K | n metal/ n AB | TOF/min -1 | E a/kJ·mol -1 | ref |
|---|---|---|---|---|---|
| PtPd cNPs | 298 | 0.002 | 50.02 | 57.3 | 152 |
| PtPd sNPs | 298 | 0.002 | 22.51 | - | 152 |
| Pt 70Ru 30-NP | 298 | 0.001 | 59.6 | 38.9 | 153 |
| PdRh-PVP | 298 | 0.003 | 1333 | 46.1 | 155 |
| Ag 1Pd 4@UIO-66-NH 2 | 298 | 0.0125 | 90 | 51.77 | 149 |
| PtNi@SiO 2 | 303 | 0.036 | 5.54 | 54.76 | 164 |
| Ni 2Pt@ZiF-8 | 293 | 0.01 | 361.4 | 23.3 | 166 |
| Pt-Co/dendrimer | 293 | 0.01 | 303 | 28.8 | 168 |
| PtCo 20/CNTs | 298 | - | 675.1 a | 42.5 | 169 |
| RuNi/TiO 2 | 298 | 0.001 | 914 | 28.1 | 173 |
| RuCo/γ-Al 2O 3 | 338 | - | 32.9 | 47 | 175 |
| RuCu/γ-Al 2O 3 | 338 | - | 16.4 | 52 | 175 |
| AuNi@MIL-101 | RT | 0.017 | 66.2 | - | 180 |
| AuCo@MIL-101 | RT | 0.017 | 23.5 | - | 183 |
| Ag-doped Ni/MIL-101 | 298 | 0.017 | 20.2 | 25 | 186 |
| AgCo/PAMAM | 298 | 0.033 | 15.84 | 35.66 | 188 |
| Pd 67Ni 33/MCN | 298 | 0.03 | 222.2 | 54.1 | 79 |
| Ni 30Pd 70/rGO | 298 | 0.01 | 28.7 | 45 | 189 |
| Ni 3Pd 7/CS | 298 | 0.024 | 187.5 | 35.32 | 191 |
| Co 35Pd 65/C | 298 | 0.024 | 22.7 | 27.5 | 192 |
| Pd@Co@MIL-101 | 303 | 0.011 | 51 | 22 | 194 |
| Fe 0.5Ni 0.5alloy | 293 | 0.12 | 11.4 | - | 202 |
| Fe 0.3Co 0.7 alloy | 293 | 0.12 | 13.9 | 16.3 | 205 |
| CuCo/graphene | 293 | 0.02 | 9.18 | - | 212 |
| Cu 0.5Co 0.5@SiO 2 | 298 | 0.08 | 4.26 | 24 | 213 |
| CuCo@MIL-101-1-U | 298 | 0.02 | 51.7 | 30.5 | 104 |
| Cu 0.2Co 0.8/PDA-rGO | 303 | 0.05 | 51.5 | 54.9 | 215 |
| Cu 0.3Co 0.7@MIL-101 | RT | 0.034 | 19.6 | - | 218 |
| Cu 0.72Co 0.18Mo 0.1 | 298 | 0.04 | 119.0 b | 45 | 222 |
| Cu 0.72Co 0.18Mo 0.1 | 298 | 0.04 | 46.0 | - | 222 |
| Cu 0.8Co 0.2O-GO | 298 | 0.024 | 70.0 | 45.5 | 237 |
| Cu 0.33Fe 0.67 | 298 | 0.04 | 13.95 | 43.2 | 223 |
| Cu 0.2Ni 0.8/MCM-41 | 298 | 0.05 | 10.7 | 38 | 224 |
| CuNi/CMK-1 | 298 | 0.072 | 54.8 | - | 226 |
aThe TOF value was calculated based on the number of Pt atom.bThe TOF value was calculated based on the reaction with the addition of NaOH (0.1 M). |
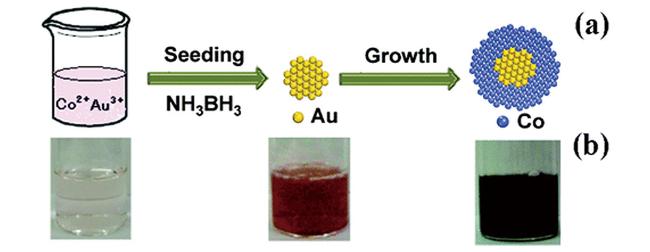
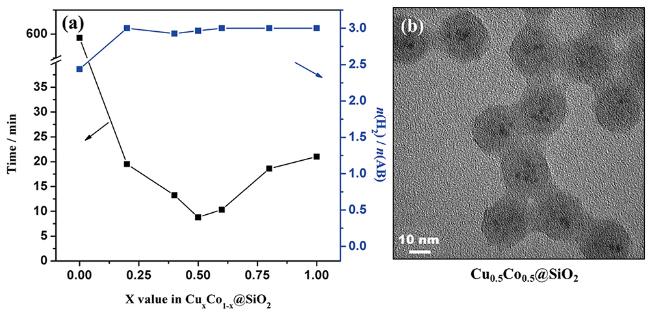
图13 (a) Cu x Co 1- x @SiO 2催化剂催化氨硼烷水解制氢性能图;(b) Cu 0.5Co 0.5@SiO 2催化剂的TEM图[213]Fig.13 (a) Cu x Co 1- x @SiO 2 core-shell nanospheres with different x values under an ambient atmosphere at 298 K;(b) TEM image of Cu 0.5Co 0.5@SiO2 [213]. Reprinted with permission from ref[213]. Copyright 2015, American Chemical Society |
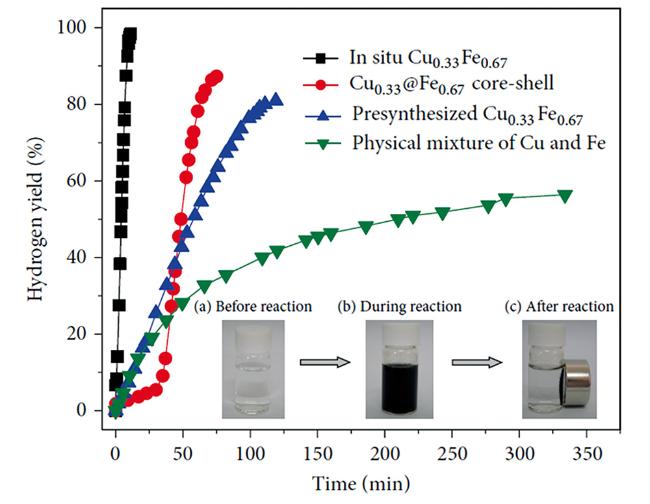
图14 不同催化剂催化氨硼烷水解制氢性能图;插图是原位合成Cu0.33Fe 0.67纳米合金的的照片[223]Fig.14 Hydrogen generation from the hydrolysis of AB in the presence of different metal nanocatalysts(metal/AB=0.04). The insert shows photographs of the catalytic hydrolysis of AB via in situ synthesized Cu 0.33Fe 0.67 nanoalloy[223]. Reprinted with the permission from ref [223]. Copyright 2013, Elsevier |
4.4 其他催化剂
5 碱对氨硼烷水解催化的促进作用
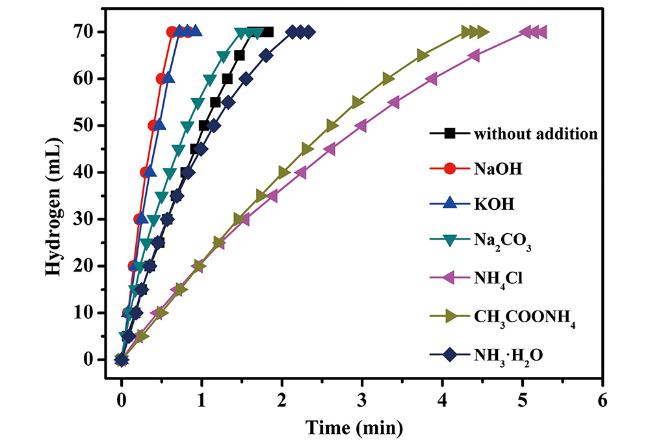
图15 Cu 0.72Co 0.18Mo 0.1催化剂在添加NaOH、KOH、Na 2CO 3、NH 4Cl、CH 3COONH 4和NH 3·H 2O条件下催化氨硼烷水解制氢性能图[222]Fig.15 Hydrogen generation from the hydrolysis of AB with the addition of NaOH, KOH, Na 2CO 3, NH 4Cl, CH 3COONH 4 and NH 3·H 2O catalyzed by Cu 0.72Co 0.18Mo 0.1 NPs at 298 K[222]. Reprinted with the permission from ref[ 222]. Copyright 2018, Royal Society of Chemistry |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}