




1 引言
硝酰氯(nitryl chloride, ClNO2)是一种无色气体,具有一定的毒性和腐蚀性,气味与氯气相似,在工业上可用作硝化剂和氯化剂[1]。ClNO2在全球氮和氯循环中起着十分重要的作用:一方面,硝酰氯是一种重要的活性含氮化合物,可在夜间临时储存NOx,并在日间通过光解将其释放至大气中以参与光化学反应和O3的形成;另一方面,ClNO2也是大气中氯自由基的重要来源,引发氯自由基的大气化学反应,从而对大气氧化性有重要贡献。
早在1989年,Finlayson-Pitts等[2]发现N2O5在NaCl表面的反应可生成ClNO2,并指出ClNO2在对流层大气中可能起到十分重要的作用。然而,受限于测量技术,之后20年并未对实际大气中的ClNO2浓度水平和大气化学行为开展深入研究, ClNO2在大气中的作用也长期被忽视。近年来,伴随着测量技术的进步,Thornton等[3,4]先后在美国沿海和内陆地区观测到了高浓度水平的ClNO2;此后,ClNO2大气化学研究逐步成为了大气化学领域的一个研究热点。本文通过总结已有的相关研究,深入探讨了大气中ClNO2的来源和去除机制、测量技术、时空分布特征及其大气环境效应,凝练了ClNO2大气化学研究中尚未解决的关键科学问题,并展望了该领域的未来发展方向。
2 硝酰氯的源和汇
2.1 来源机制
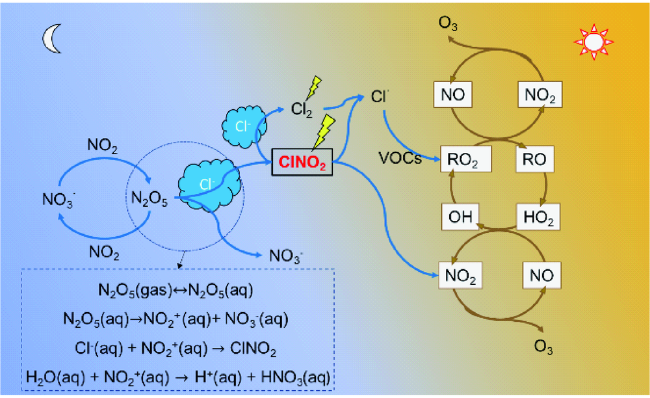
目前我们已经认识的ClNO2大气化学反应框架如图1所示。ClNO2主要来源于N2O5与含氯气溶胶或者地表的非均相反应(R1),其中Φ为ClNO2的产率。N2O5通过扩散、传质、溶解和反应在颗粒物中生成NO2+和NO3-;NO2+可与颗粒物中的Cl-反应生成ClNO2,也可与颗粒物中的H2O反应生成NO3-[5]。由于NO3在日间快速光解且可被NO快速滴定,且N2O5和NO3之间存在快速的热力学平衡,所以日间NO3和N2O5的浓度水平较低,因此一般认为NO3和N2O5的大气化学反应主要发生在夜间[6];而ClNO2通常在夜间由N2O5非均相反应生成并逐步积累[5,7~9]。此外, Cl自由基与NO2的反应也可生成ClNO2(R2),该反应的速率常数为1.48×10-30 cm-6· molecule-1·s1(296 K)[10];由于实际大气Cl自由基的浓度很低(103~106 molecule·cm-3)[11],该反应(R2)对ClNO2的贡献可忽略。
2.2 去除机制
ClNO2在夜间形成和积累,然后在日间发生光解并产生Cl自由基和NO2。Cl自由基活性非常强,和大部分烷烃类物质的反应速率远超OH自由基的反应速率。Cl自由基和挥发性有机物(VOCs)的反应生成RO2,从而与ClNO2光解产生的NO2共同影响日间光化学。现有研究表明,ClNO2的光解是对流层大气Cl自由基的重要来源,在区域和全球尺度上影响大气氧化性以及活性含氮物种的大气寿命,并促进O3和RO2的生成,加剧日间光化学污染[4,13]。另外,N2O5的非均相反应是硝酸盐气溶胶的重要来源[14];而ClNO2是该非均相反应的重要产物之一。所以,Φ(ClNO2)在很大程度上决定了硝酸盐和ClNO2的生成,是准确模拟和评估夜间非均相化学对O3和PM2.5贡献的关键参数之一。
在中性或者弱酸性条件下,气溶胶与ClNO2的非均相反应较慢;但当颗粒物pH值小于2时,ClNO2和Cl-发生酸催化反应快速生成Cl2(R4)。 实验室研究[15]发现,酸催化条件下ClNO2的摄取系数能够达到10-3量级,这表明该反应可能是大气中Cl2的重要来源,能进一步实现大气的氯活化。但是,最新的一项外场观测研究(Haskins等, 2019)认为[76],实际大气条件下酸催化过程并不重要,该反应的摄取系数可能在10-6 ~ 10-5量级,远远低于实验室结果。这可能是因为实验室研究中NaCl样品的厚度较薄,非均相反应未受液相扩散的限制;而对于实际大气颗粒物,ClNO2将主要与颗粒物内部的Cl-反应,导致该非均相反应被液相扩散限制。
$ClNO_{2}+Cl^{-}(aq)\to Cl_{2}+NO_{2}^{-}(aq)$
ClNO2+ OH·→ HOCl + NO2
k (ClNO2+OH)=2.4×10-12∙exp(-1250/T) (260 ~ 350 K)
3 测量技术
3.1 直接测量技术
早期研究主要使用红外光谱来测量ClNO2[18],如利用ClNO2在7.9和12.6 μm附近的红外特征吸收对其进行定性和定量分析。但是该方法的灵敏度有限,通常只用于实验室研究,难以检测实际大气中的ClNO2。
化学离子化质谱技术(CIMS)是目前测量实际大气中ClNO2的主要技术,其基本原理是采用碘离子(I-)与ClNO2反应,生成I(ClNO2)-和ICl-(R6,R7)。
I-+ClNO2→I(ClNO2)-
I-+ClNO2→ICl-+ NO2
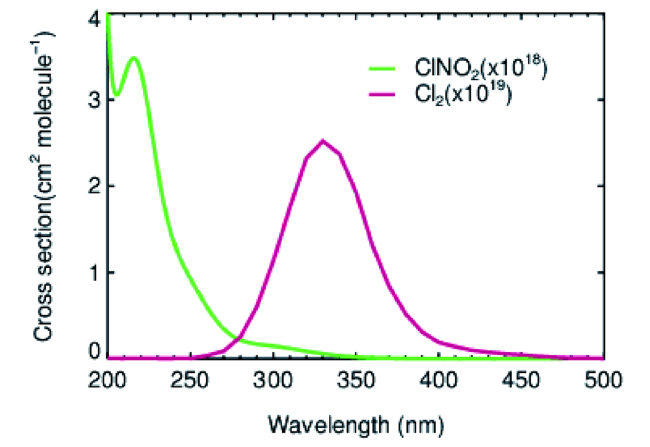
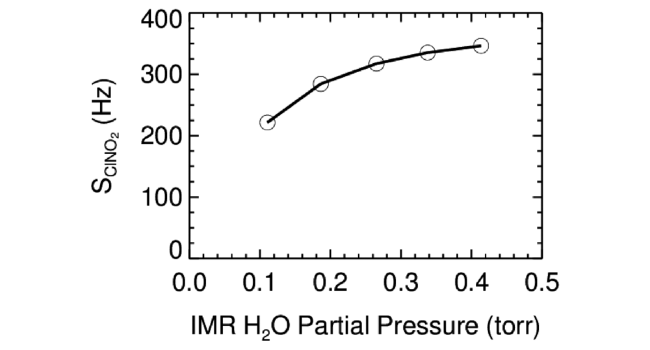
35Cl和37Cl的环境丰度比值为3.13∶1[19],因此在m/z为163.9和161.9 amu [7,20]或者在207.9和209.9 amu处可测量到比例恒定的ICl-或I(ClNO2)-质谱信号[21,22]。早期研究通过测量ICl-来定量ClNO2的浓度,后续研究发现HCl可能对ICl-测量存在一定干扰[20]。目前主要利用I(ClNO2)-来定量测量ClNO2的浓度,其主要原因是特异性较好、干扰相对较小且背景值低。该方法在一定程度上会受到HNO3·H2O的影响,采用高分辨质谱能够有效分离质谱峰以排除干扰,但是低分辨的四级杆质谱可能会导致测量结果偏高[23]。如图3所示,H2O可以与I-结合成I(H2O)-,该离子继而与ClNO2反应结合生成I(ClNO2)-,所以H2O的存在将影响(一般是促进)I(ClNO2)-的形成。因此,实际测量需要定量考虑大气中的水蒸气含量对仪器灵敏度的影响。目前CIMS测量ClNO2的检测限通常为1~10 pptv,不确定性一般为20%~40%,基本能够满足外场观测的需求[21,22,24,25]。
3.2 间接测量技术
3.3 硝酰氯测量的标定
对仪器信号进行标定是使用CIMS测量ClNO2的关键技术难点之一。由于目前没有商业化的ClNO2标准源,一般采用自行研发ClNO2标准源来对测量系统进行标定,而目前常用的标准源主要有两种。第一种是将Cl2通入到湿润的NaNO2粉末中,两者将发生反应并生成ClNO2和NaCl(R8),然后通过CRDS技术来测量ClNO2 热解生成的NO2。CRDS测量NO2具有较好的检测限(<0.1 ppbv),ClNO2源产生的ClNO2浓度通常在ppbv量级,采用CRDS完全能够保证对ClNO2源浓度的准确定量;为了抑制NaNO2分解生成NO2,通常在NaNO2中加入少量NaCl。第二种方式是将N2O5通入到湿润的NaCl粉末中,两者发生反应并生成ClNO2和NaNO3 (R9);该反应中ClNO2的产率为100%,通过测量N2O5的消耗便可获得ClNO2的浓度[2,7,27]。目前,第二种方法的应用更为广泛[22]。此外,ClNO2也可通过NOCl和O3的反应[28]或者硝酸钾和无水三氯化铝的反应[29]来合成,但是这些合成方法尚未应用于大气化学研究中。
Cl2+NO2- → ClNO2 + Cl-
NaCl+N2O5 → ClNO2 + NaNO3
4 硝酰氯的分布特征和环境影响
4.1 时空分布特征
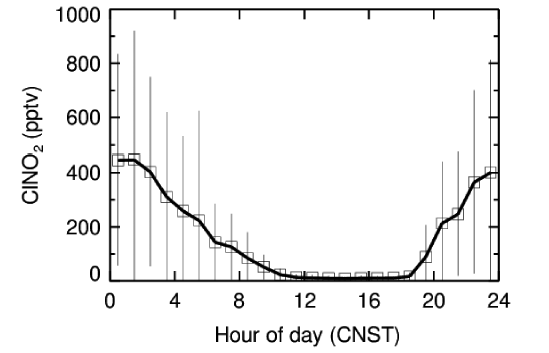
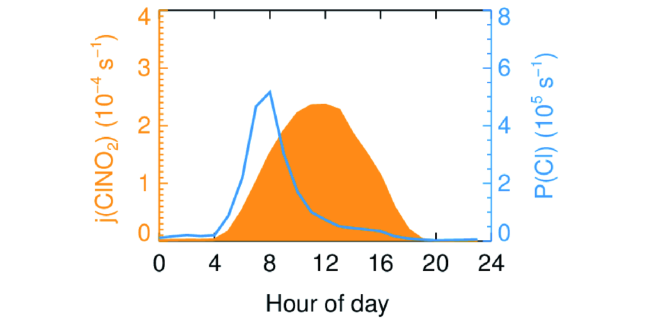
近年来,CIMS技术在全球各种不同环境条件下被用于ClNO2的外场观测。表1总结了目前国内外外场观测所报道的ClNO2浓度水平和N2O5在颗粒物表面生成ClNO2的产率。ClNO2浓度通常在几十pptv至几个ppbv量级,目前所报道的最高浓度为4.7 ppbv(1 min)[13]。ClNO2的外场测量研究已经覆盖了城市、郊区以及沿海地区,观测形式也从近地面测量发展到了塔基测量、船舶测量以及飞机航空测量。图4为北京地区夏季ClNO2日变化结果,代表实际大气中的典型日变化:日落之后N2O5的非均相反应逐渐生成ClNO2并持续积累,次日日出之后由于光解,其浓度将降低,高于检测限度的ClNO2能够持续到中午甚至下午。部分外场观测发现ClNO2在清晨出现峰值,这可能是由高空垂直向下输送所导致的[24,30~32]。
表1 文献报道的国内外外场观测ClNO2浓度以及ClNO2产率的结果汇总Table 1 Summary of the results of field observations of ClNO2 concentration and ClNO2 yield reported in the literatures |
| Location | Region | ClNO2 (ppb) (max, mean) | ClNO2 yield (range, points) | ref |
|---|---|---|---|---|
| Wangdu, CN | Rega | 2.07, 0.55 | 0.06~1.04 (10) | 24 |
| Jinan, CN | Urban | 0.78, 0.09±0.06 | 0.01~0.08 (4) | 37 |
| Mt Tai, CN | RLb | 2.1, 0.05±0.11 | 0.17~0.90 (8) | 46 |
| Beijing, CN | Urban | 1.4, 0.17±0.26 | 0.10~0.35 (4) | 36 |
| HK, CN | Coastal | 4.7, n.a. | 0.02~0.98 (9) | 47 |
| Beijing, CN | Rural | 2.9, 0.8 | 0.5~1.0 (5) | 48 |
| Seoul, KOR | Urban | 2.5, n.a. | n.a. | 49 |
| Seoul, KOR | Rega | 0.8, n.a. | n.a. | 49 |
| London, UK | Urban | 0.72, n.a. | n.a. | 31 |
| Calgary, CA | Rural | 0.34, 0.04 | 0.005~ 0.12 | 25 |
| KFd,GER | Rural | 0.7, n.a. | 0.035~1.38 (33) | 39 |
| North Coast, UK | Coastal | 0.065, 0.01 | n.a. | 40 |
| BC, Canada | Coastal | 0.1, 0.008 | 0.7 (1) | 50 |
| UK | Lc | 4.1, 0.9 | n.a. | 41 |
| Mediterranean | Coastal | 0.439, 0.02 | 0.53 (1) | 38 |
| Suez | Coastal | 0.586, 0.075 | 0.90 (1) | 38 |
| Red Sea | Coastal | 0.480, 0.047 | 0.86 (1) | 38 |
| Aden | Coastal | 0.379, 0.041 | 0.76 (1) | 38 |
| Arab. Sea | Coastal | 0.056, 0.007 | 0.87 (1) | 38 |
| Oman | Coastal | 0.376, 0.067 | 0.50 (1) | 38 |
| Arab. Gulf | Coastal | 0.126, 0.021 | 0.17 (1) | 38 |
| Houston, US | Coastal | 1.2, n.a. | 0.1~ 0.65 (2) | 3 |
| Boulder, US | Rural | ~0.45, n.a. | n.a. | 4 |
| LA, US | Urban | 2.15, n.a. | n.a. | 43 |
| Boulder, US | Rural | 1.3, 0.27 | 0.45~0.8 (2) | 42 |
| Weld, US | Rural | n.a., 1.2±0.7 | 0.05~0.90 (85,440) | 51 |
| Calif, US | Urban | 3.6, n.a. | n.a., n.a. | 44 |
| Houston, US | Urban | 0.1, n.a. | n.a. | 32 |
| Eastern, US | RLb | n.a., n.a. | 0.003~1 (3425) | 52 |
*aReg: Regional;bRL: Residual layer;cL: Landfill;dKF: Kleiner Feldberg |
高浓度的ClNO2通常出现在受人类活动影响的沿海地区以及城市下风向地区。在海洋背景地区,ClNO2的浓度通常低于0.1 ppbv[3]。ClNO2的生成主要受三方面的影响。首先是N2O5生成,人为活动影响的区域NOx排放量大,N2O5的浓度较高。其次是颗粒物中氯的含量,在海洋及沿海地区易出现高浓度的ClNO2,内陆地区的Cl源则主要包括生物质和煤炭燃烧以及冬季撒盐融雪等人为活动[33];最新研究表明,含氯矿尘颗粒物也可能是内陆地区ClNO2的一个潜在来源[9]。最后是N2O5非均相反应速率的限制,不同类型气溶胶表面的N2O5非均相反应速率差异可以高达多个量级[34,35],同时还受到颗粒物表面积的影响,因此在城市下风向地区高颗粒物浓度条件下,容易出现高浓度ClNO2。现有的外场观测表明,我国城市地区(如华北和珠三角地区)ClNO2的浓度水平普遍可达到ppbv量级[13,24,30,36,37]。船舶航测表面,地中海地区ClNO2浓度处于中等水平,最高浓度水平不超过0.6 ppbv[38];此外,德国和英国近地面观测的ClNO2浓度水平也基本相当[31,39,40]。最近Bannan等[41]发现垃圾填埋场附近ClNO2浓度可以高达4 ppbv,表明垃圾处理过程是潜在的氯活化重要来源。我国垃圾处理总量高,垃圾填埋区也可能是ClNO2的热点地区,可能对附近城市地区的光化学过程产生一定影响。北美是最早开始开展ClNO2观测研究的地区,近年来开展了一系列ClNO2外场观测,其ClNO2浓度水平与我国城市地区大体相当[3,4,42~44]。长期观测显示,加拿大阿尔伯塔地区夜间积累的ClNO2呈现夏季低冬季高的特征[25],欧洲地区的长期连续观测也发现同样的季节变化特征[45]。ClNO2浓度分布的季节差异原因在于一方面冬季夜间时长较夏季长,能够提供更长的反应时间,另一方面可能是冬季低温导致NO3-N2O5化学热力学平衡趋向于N2O5化学,且冬季颗粒物组分(如氯离子)较夏季有所区别,可能导致N2O5非均相反应具有相对较高的ClNO2产率,目前在ClNO2浓度水平的季节调控原理方面的研究还不全面。但总的来看,冬季OH自由基的浓度水平相对较低,但ClNO2贡献的Cl自由基在冬季有所增加,侧面反映了ClNO2化学在冬季可能更为重要[25];但是,目前ClNO2的外场观测主要集中在夏季,冬季观测则相对较少。
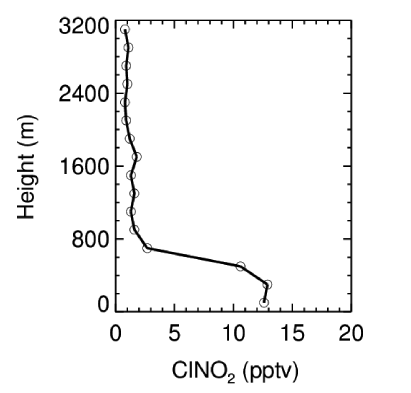
目前已有的ClNO2外场观测大部分是地面观测,关于ClNO2垂直分布的观测研究非常有限。Young等[53]首次报道了美国加州地区ClNO2航测研究结果,发现ClNO2在夜间边界层内浓度水平高且垂直分布较均匀,但在残留层浓度陡降。在韩国和英国航测中得到的ClNO2垂直分布趋势和美国的航测结果基本一致[40,49],但是在英国航测中观测的ClNO2在整个垂直尺度上的浓度水平非常低,如图5所示[40]。此外,美国博尔德地区的塔基观测(0~300 m)发现, ClNO2的近地面浓度较低,而在100~300 m高度范围内浓度较高,主要决定于不同高度下NO2的浓度水平[42]。近年来在美国开展了更多的ClNO2航测研究,如2015年美国的WINTER航测实验,但该观测中的ClNO2的浓度水平及其垂直分布特征尚未见发表[54]。
4.2 硝酰氯的非均相生成和产率
近年来,包括美国国家大气海洋局、华盛顿大学、德国马克斯-普朗克研究所、英国曼彻斯特大学、香港理工大学、山东大学、中国科学院大气物理所和北京大学等开展了系列关于ClNO2非均相生成的实验室、外场和模式研究。然而,目前关于ClNO2非均相生成的反应机制以及相关影响因素研究的科学认识还不够深入。
4.2.1 实验室研究
在N2O5与气溶胶非均相反应并生成ClNO2的过程中,N2O5的摄取系数和ClNO2的产率是两个关键参数。其中N2O5的摄取系数受到气溶胶理化性质和环境条件的影响,如颗粒物含水量、有机物、硝酸盐、氯盐以及温度和相对湿度等[34]。但是,很多外场观测和数值模拟研究发现基于实验研究结果的N2O5摄取系数参数化方法往往高估了实际大气中N2O5的非均相摄取[46,52,55,56]。近期Yu等[57]通过对香港理工大学所开展的N2O5和ClNO2外场观测的全面分析,修正了Bertram和Thornton提出的经典参数化方案[5],并发现修正后的参数化方案可以更好地预测实际大气中N2O5的摄取系数。虽然暂时不清楚Yu等提出的新参数化方案是否也适用于其他地区,但是至少该参数化方案可以较好地模拟中国东部地区N2O5的非均相摄取。
实验室研究一般通过测定控制条件下由于非均相反应造成的N2O5的消耗量(Δ[N2O5])和ClNO2的生成量(Δ[ClNO2])来测定ClNO2的产率,即Φ(ClNO2)(Eq. 2):
NO2+(aq)+H2O(aq)→NO3-(aq)+2H+(aq)
NO2+(aq)+Cl-(aq) →ClNO2
如图6所示,实验室研究表明,在N2O5与大气颗粒物的非均相反应中,Φ(ClNO2)随着颗粒物中Cl-浓度增加而逐步上升。假设Φ(ClNO2)仅受氯离子和颗粒物含水量这两个影响因素控制,则可以使用Eq. 2对Φ(ClNO2)进行参数化[27]。由于目前还无法测定k10a和k10b,只能通过拟合ClNO2产率与[H2O/[Cl-]的关系来获得k10b/k10a。早期Behnke等所测定的k10b/k10a值为836±32[27],而Bertram和Thornton所测定的k10b/k10a值为483±175[5],远低于Behnke等的测量结果。此外,Roberts等所测定的k10b/k10a值为450±100[8];考虑到测量误差,Roberts等的研究结果与Bertram和Thornton的研究结果非常一致。后续的外场观测和数值模拟研究普遍使用Eq. 2来模拟ClNO2的非均相生成,所采用的k10b/k10a值一般为483或450。
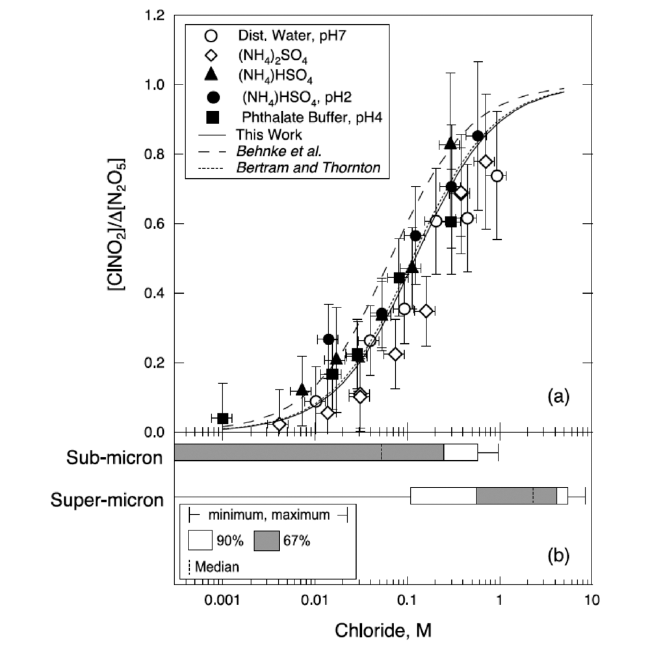
图6 (a)实验室定量的ClNO2产率与颗粒物中Cl-物质的量的响应关系(摘自文献[8]图3, 版权归属于John Wiley and Sons);(b)2016年墨西哥湾TexAQS-GoMACCS外场观测期间测得的亚微米和超微米颗粒物中氯离子的浓度范围Fig.6 (a) The efficiency of conversion of N2O5 to ClNO2 as a function of substrate chloride concentration, and (b) the range of sub- and super-micron chloride concentrations measured during the TexAQS-GoMACCS 2006 field study in the Gulf of Mexico (original figure referred from[8], copyright: John Wiley and Sons) |
4.2.2 外场观测及数值模拟验证
第二种方法是产物拟合方法,该方法基于观测到的N2O5浓度和颗粒物表面积浓度来计算ClNO2和硝酸盐的增长趋势,并通过调整γ(N2O5)和Φ(ClNO2)来优化计算结果,直到计算的ClNO2和硝酸盐浓度增长和观测结果吻合;该方法同样只适用于ClNO2和硝酸盐同时增长的过程[39,46]。第三种方法是迭代盒子模型模拟法,目前主要应用在夜间残留层的环境条件,基本原理如下:假设夜间过程采集到的每一个数据点都是单独的气团,气团反应起始时间设置为日落或日落前的某个特定时间点,此时N2O5和ClNO2的浓度为零;随后气团开始夜间化学反应过程,反应时长为起始时间点至测量时间点,该反应过程包含一个简单的化学体系,即NO2和O3生成NO3和N2O5,N2O5的非均相反应主导NO3的去除;通过盒子模型先后迭代反应起始点的NO2和O3浓度和测量时间点的N2O5浓度,不断调整γ(N2O5)直到模拟的N2O5浓度和观测时间点的浓度吻合,随后通过限定N2O5摄取系数进一步迭代Φ(ClNO2)直到模拟的ClNO2浓度和观测值吻合,即得到了观测时间点对应的Φ(ClNO2)[51,54]。相比前两种方法,第三种方法的适用范围更广,但是需要系统评估排放气团注入和气体沉降等多方面因素对迭代结果的影响;同时,该方法的前提假设较多,具有不确定性。表1总结了目前外场观测所报道的Φ(ClNO2)。可以看出,在不同的环境条件下,Φ(ClNO2)具有很大的差异。
很多外场观测研究[42,46,51,54,56,61]发现,目前应用最为广泛的参数化方案(Eq. 2)普遍高估了Φ(ClNO2)。在2010年5~6月的美国加州观测中,Mielke等[44]发现把k10b/k10a下调至50后,Φ(ClNO2)的计算值将与观测值更为接近。Tham等[56]发现对于我国望都夏季,所观测到的Φ(ClNO2)与颗粒物中氯离子浓度没有正相关关系,反而与含水量有一定的正相关,这与实验室结果存在一定矛盾;此外,McDuffie等通过分析2015年春季美国飞机航测的数据,也发现Φ(ClNO2)和含水量正相关这一和实验室结果相反的现象[54]。上述外场观测结果表明,现阶段的参数化方法(Eq. 2)不能准确描述实际大气中的Φ(ClNO2),其可能原因包括:1)实际大气颗粒物中可能还存在H2O和Cl-之外的能与NO2+反应的反应物; 2)实际大气条件下颗粒物中Cl-的活度大大低于其浓度; 3)在实际大气中存在着目前我们还未认识到的ClNO2去除途径。
近期Yu等[57]分析了香港理工大学所开展的N2O5观测,修正了γ(N2O5)参数化方案,并发现修正后的参数化方案可以更好地预测实际大气中的γ(N2O5)。基于修正后的γ(N2O5)参数化方案,可以得出k10b/k10a的值应为105±37,即:
该方案与目前通用的Φ(ClNO2)参数化方案相比,修正后的参数化方案(Eq. 4)可以更好地估算实际大气中的Φ(ClNO2);另一方面,Eq. 4仍然高估了实际大气中的Φ(ClNO2),且不能解释实际大气条件下Φ(ClNO2)的变化幅度。考虑到大气颗粒物中的有机物抑制ClNO2的生成,我们提出了一个新的参数化方案,以考虑有机物对Φ(ClNO2)的影响:
其中fOA为大气颗粒物中有机组分的质量分数。可以预见,Eq. 5将进一步降低Φ(ClNO2)的计算值,从而可能使之与观测值更为接近,但是Eq. 5 的具体效果还有待实际观测来验证。此外,不同的有机物对ClNO2生成的抑制作用存在差异;但是由于相关科学认识的缺乏,目前Eq. 5暂时未考虑不同成分的有机物对Φ(ClNO2)的影响。
目前我们尚不清楚导致Φ(ClNO2)的观测值显著高于参数化方案计算值的原因。Tham等[56]发现老化气团的Φ(ClNO2)较新鲜排放气团高,且Φ(ClNO2)和CH3CN/CO比值呈反相关,推测Φ(ClNO2)的高估可能和颗粒物中的有机物有关。实验室研究[61,62]也初步证实了颗粒物中有机物可能和NO2+竞争气溶胶中的Cl-,从而抑制ClNO2的生成并降低Φ(ClNO2)。然而,实际大气气溶胶中的有机物构成复杂,目前还缺乏不同有机物对ClNO2生成的抑制效果以及其机理的研究。考虑到ClNO2在大气化学中的重要角色,Φ(ClNO2) 的准确参数化能有效提高对O3、硝酸盐以及相关污染物的模拟和预测,因此Φ(ClNO2)是目前研究尚待解决的关键问题。
4.3 硝酰氯化学的环境效应
4.3.1 对氯自由基收支的影响
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
表2 已有观测中ClNO2贡献Cl·生成速率日间峰值汇总Table 2 Summary of the campaign average daily peak of the chloride radical via ClNO2 photolysis reported in previous filed campaigns |
| Location | Region | P(Cl·) (×105 cm-3·s-1) Mean diurnal peak | ref |
|---|---|---|---|
| Hong Kong, CN | Coastal | 3.1 | 64 |
| Wangdu, CN | Regional | 16.3 | 24 |
| Beijing, CN | Rural | 5.2 | 30 |
| Hessen, GER | Rural | 12 | 63 |
| London, UK | Urban | 2.5 | 31 |
| Calgary, CA | Rural | 7 (Campaign peak) | 25 |
| Norfolk Coast, UK | Coastal | <1.0 | 40 |
| Houston, US | Urban | 8.0 | 3 |
| Boulder, US | Rural | 2.5 (Estimated) | 4 |
| Los Angeles, US | Urban | 25 | 53 |
| Houston, US | Urban | 3.4 (Estimated) | 32 |
4.3.2 对大气氧化性和臭氧生成的影响
当前我国面临着严重的复合型大气污染,O3和PM2.5已经成为了大气污染防治和空气质量管理的两大难题。Cl自由基氧化VOCs后生成RO2自由基,RO2进一步参与光化学循环反应从而促进HOx自由基和O3的生成;与此同时,ClNO2光解产生的NO2也将参与光化学反应并影响O3的生成。ClNO2化学将提高模拟的HOx自由基和O3的浓度水平,在一定程度上弥补日间自由基浓度以及O3生成速率的观测值和模拟值之间的差异,因此在O3生成和自由基化学中可能扮演着重要的角色。
研究表明,美国加州地区ClNO2化学对日间O3生成的贡献可达12 ppbv[65]。在我国华北地区,ClNO2化学对早晨初级RO2来源贡献可达10%~30%,在污染过程中对O3生成量的贡献达11 ppbv,其贡献率则高达13% [24]。在我国香港地区,ClNO2对O3生成贡献可达11%~41%,高浓度的ClNO2可额外增加77%的OH和106%的HO2生成;然而,不同气团中ClNO2浓度差异较大,所以ClNO2对大气氧化性贡献也具有较大的不确定性[13]。区域模型研究表明,ClNO2将有效增加日间O3的初级来源[65,66,67],其中日间最大8 h O3浓度增加量最高可达7 ppbv[68]。以我国华北地区为例,在CMAQ中加入ClNO2的非均相生成等机制后,Cl化学对部分区域的O3以及自由基贡献分别可达20%以及28%~48%[69]。
4.3.3 对硝酸盐生成的影响
早期模型研究中尚未考虑ClNO2的非均相生成,因此硝酸盐是模型中N2O5非均相反应的唯一产物,导致模拟的硝酸盐浓度偏高。在考虑了ClNO2的非均相生成之后,实质上也降低了N2O5的非均相反应对硝酸盐生成的贡献;另一方面,ClNO2的光解将提高NO2和HOx自由基的浓度,从而增强OH与NO2的反应对硝酸盐形成的贡献。
5 结论与展望
过去二十年中,国内外实验室研究、外场观测和模式模拟的广泛开展大大提高关于ClNO2浓度分布特征、化学机理及其大气化学意义的科学认识。相关研究领域的主要成果包括:
(1)开发并优化了基于CIMS技术测量ClNO2的方法,并建立了相对完善的数据质量控制方案,使得我们能够有效开展实验室研究以及在全球的不同环境条件下开展近地面测量、船舶测量以及飞机航测等。
(2)发现ClNO2在海洋及沿海地区、城区、郊区以及背景地区等广泛存在,在一定程度上揭示了ClNO2的典型浓度水平和时空分布特征,为准确评估ClNO2在大气环境中的作用提供了基础数据。
(3)发现气溶胶中液态水、氯离子和有机物等影响ClNO2的非均相生成,并基于实验室研究结果提出了相应的参数化方案,但是目前已有的参数化方案普遍高估了实际大气中ClNO2的非均相生成。
(4)指出ClNO2的光解是对流层大气中重要的氧化剂和自由基来源,对一次污染物的降解和二次污染物的形成具有重要贡献,深化了我们对大气自由基化学的认识。
近年来我国大气污染防治也取得了一系列重要成果, 空气质量得到了持续改善。随着我国减排措施的有效实施,SO2减排效果显著,颗粒物中硫酸盐比重显著下降。但是,我国PM2.5浓度仍然远高于世界卫生组织指导值,进一步减排的难度增大,且冬季硝酸盐的质量浓度和相对贡献突出;与此同时,我国臭氧污染问题也日趋严重。为了更有效地解决我国大气复合污染,在大气污染成因研究上需要着重阐明气相和非均相过程的霾化学机理,在空气污染治理上则需要进一步强调协同减排和联防联控联合发展[72]。N2O5的非均相摄取和ClNO2的非均相生成,不但耦合了气相化学与非均相化学,而且也耦合了夜间大气化学与日间光化学过程,在大气化学研究尤其是霾化学研究中具有非常重要的地位。现阶段ClNO2大气化学研究仍然面临诸多科学难题,亟待开展的进一步研究主要包括:
(1) 丰富大气中硝酰氯的在线测量技术和测量手段。开发不同原理的ClNO2测量系统,开展不同原理的测量技术比对,提高ClNO2测量准确性和测量系统的便携性。
(2)开展典型大气环境条件下ClNO2的连续在线观测研究,积累实验室研究和模型模拟的验证和比对的数据样本,尤其是不同环境条件下的长时间连续在线监测和垂直廓线测量研究。
(3)目前在全球内陆地区均观测到了高浓度的ClNO2,但是颗粒物中氯离子的来源还不完全清楚。我国已经开展了一些氯排放清单的工作,后续需要进一步在内陆地区开展亚微米级颗粒物中氯离子来源的调查研究,并完善我国氯排放清单[73]。
(4)定量解析ClNO2的来源去除机制,重点解析N2O5非均相反应机制,ClNO2产率的关键影响机制以及ClNO2自身的非均相反应等,结合观测和实验对上述反应的关键反应过程和参数进行准确定量和参数化,定期更新模型机理并结合观测数据优化模型表现[74]。
(5)开展氯自由基大气化学的研究,如氯自由基和VOC的反应动力学、反应机理开发[75]以及氯化学对二次有机气溶胶生成贡献研究等,以准确评估卤素化学在区域和全球大气尺度下的大气化学作用。