



1 引言
2 实验方法简介
2.1 二维相干红外光谱

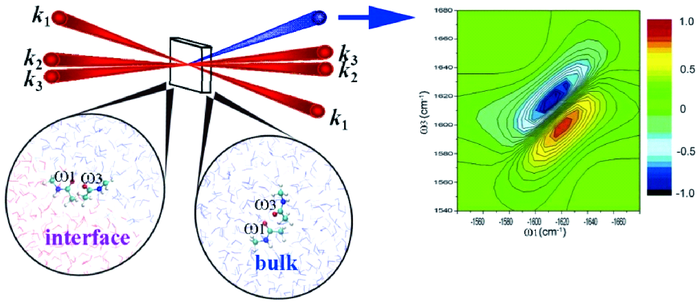
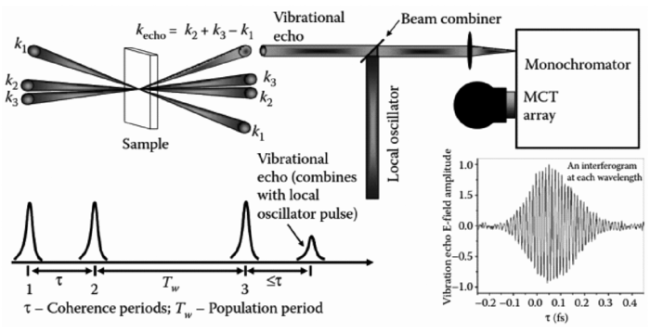
图2 外差式探测(heterodyne-detected)的二维相干红外光谱实验示意图(左): 三个时间延迟t1, t2, t3 保持第三个时间延迟固定,并在涉及两个时间延迟的演化得到二维相关信号图。(右上): 沿相位匹配方向; $k_{I}=k_{1}+k_{2}+k_{3}$的两个耦合振动的二维光子回波谱。 ω1 和ω3是t1 和t3的傅里叶共轭物理量。 两种振动模式的频率涨落分别是慢速和反相关(左),慢速和相关(中),快速和反相关(右)。(右下):三个模型对应的线性吸收光谱Fig.2 (left) Pulse configuration for a heterodyne detected multidimensional four-wave mixing experiment. Signals are recorded versus the three time delays t1, t2, t3, and displayed as 2D correlation plots involving two of the time delays, holding the third time delay fixed;(right top): 2D photon-echo spectra of two coupled vibrations in the phase-matching direction $k_{I}=k_{1}+k_{2}+k_{3}$. ω1 and ω3are the Fourier conjugate variables to t1 and t3. The frequency fluctuations of the two modes are slow and anti-correlated(left panel), slow and correlated(middle panel), fast and anti-correlated(right panel).(Right bottom): Linear absorptions for the three models |
2.2 界面和频振动光谱(SFG-VS)
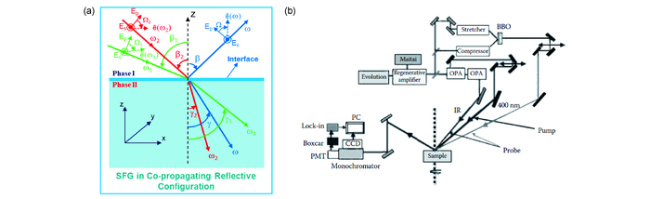
图4 (a)和频振动光谱基本原理示意图[35]。(b)时间分辨SFG实验中使用的实验装置示意图。设置用于研究表面及界面的过程的超快光谱和界面分子结构,及溶剂化、相互作用、能量和电子转移动力学等。其中泵浦光(pump)可以沿表面法线入射,也可以由其他角度入射[28]Fig.4 (a) Basic principle of sum-frequency generation vibrational spectroscopy[35]; (b) Schematic diagram of experimental device used in time-resolved SFG experiment. The experimental device is configured to study ultrafast spectrum and molecular structure of the interface, solvation, interaction, energy and electron transfer dynamics, etc. of the surface and interface processes. The pump can be incident along the normal of the surface or other angles[28] |
3 溶液相振动光谱的动力学理论描述
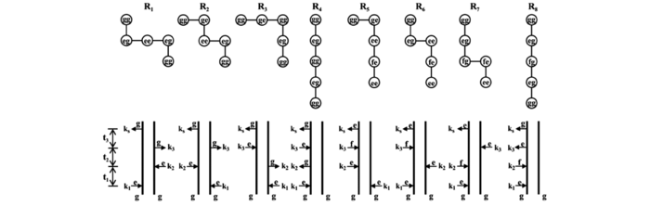
图5 常见的八条刘维尔空间路径(上)及其对应费曼图(下),每条刘维尔空间路径从左上的圆开始,而费曼图从底部开始演化[51]Fig.5 Eight Liouville-space pathways(upper) and double-sided Feynman diagrams(lower) for two-dimensional spectroscopy in the rotating wave approximation. Each Liouville-space pathway starts from the upper-left circle, while the double-sided Feynman diagram starts from the bottom[51] |
3.1 SOS(sum over-states)方法
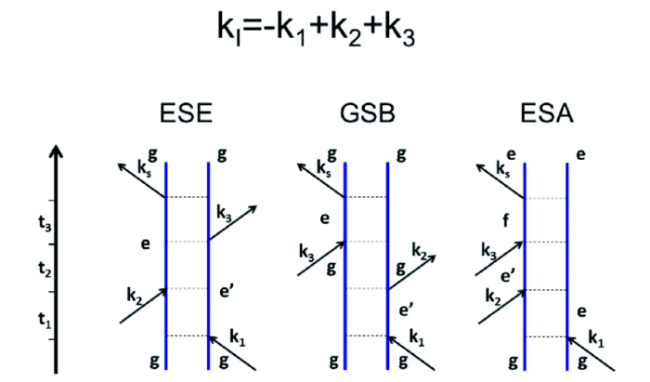
图6 表示在旋转波近似中有助于kI信号的Liouville空间路径的费曼图。这三种途径被称为激发态发射(ESE),基态漂白(GSB)和激发态吸收(ESA)Fig.6 Feynman diagram representing the Liouville space path for the kI signal in the rotating wave approximation where these three pathways are called excited state emission(ESE), ground state bleaching(GSB), and excited state absorption(ESA)[32] |
3.2 NISE(numerical integration of the Schrödinger equation)方法
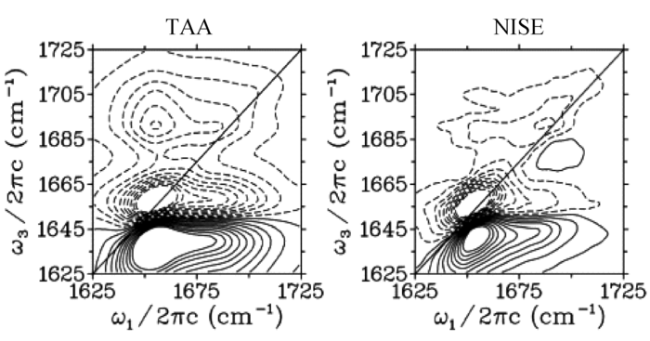
3.3 NEP(nonlinear exciton propagation)方法
3.4 SLE(stochastic Liouville equations)方法
4 2DIR技术及其在溶液化学中的应用
5 和频振动光谱技术及其在表面和界面化学中的应用

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
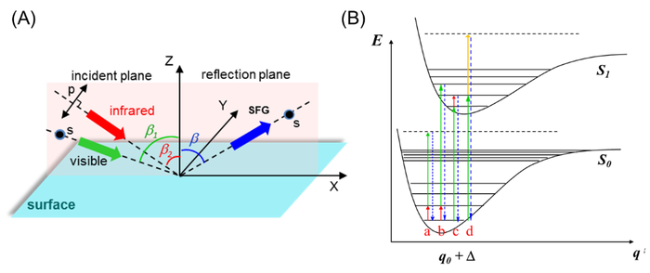
图9 和频振动光谱原理示意。(A)共传播构型下的和频振动光谱的ssp偏振组合中和频振动光谱信号、入射的可见和红外光处于与界面垂直的共同入射平面中,其中光场的偏振方向p是指光电场方向在入射平面内,而偏振方向s是指光电长矢量方向与入射面垂直;(B)和频振动光谱的不同形式。(a)单共振和频振动光谱,基态振动态共振;(b)红外+可见双共振和频振动光谱,基态振动态和电子激发态双共振; (c)可见+红外双共振和频振动光谱,电子激发态和激发态振动态双共振;(d)可见+可见双共振和频电子光谱。(b、c、d)三种情形可以用于研究电子激发态相关的表面界面分子结构、传能及电子与质子转移过程等[36]Fig.9 Illustration of the SFG-VS.(A) Showing the infrared(IR) and visible(Vis) beams incident in the same plane in a co-propagating geometry, with the SFG signal, the visible beam and the IR beam in the ssp polarization combination. The polarization of the optical field is defined as p polarization when the optical filed vector is within the incident plane, and it is s polarization when the optical field is perpendicular to the incident plane.(B) Various types of SFG processes that can be used to study ground state vibrational spectra and electronic spectra of molecular surfaces and interfaces. Type (a) is called the IR-SFG-VS, the most common single resonance SFG-VS with only the IR frequency in resonance with the ground state vibrational transition of the molecule of interest; Type (b) is called the IR-Vis double resonance SFG-VS(IR-Vis SFG-VS), where the IR frequency is in resonance with the ground state and the visible is in resonance with the electronic resonance of the molecule; Type (c) is called the Vis-IR double resonance SFG-VS(Vis-IR DR-SFG-VS); and the Type (d) is the Vis-Vis SFG-VS[36] |