




Contents
1 Introduction
2 UDA tandem reaction with furan as diene
3 UDA tandem reaction with pyrrole as diene
4 UDA tandem reaction with thiophene as diene
5 UDA tandem reaction with oxazole as diene
6 UDA tandem reaction with 1,2,4-triazine as diene
7 UDA tandem reaction with benzene as diene
8 UDA tandem reaction with unsaturated bond and aromatic ring as diene
9 Ugi/retro Diels-Alder tandem reaction
10 Conclusion
1 引言
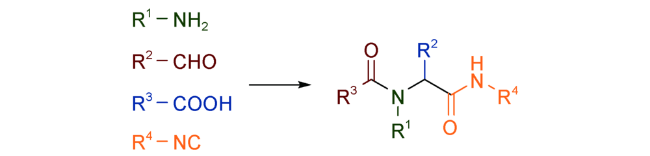
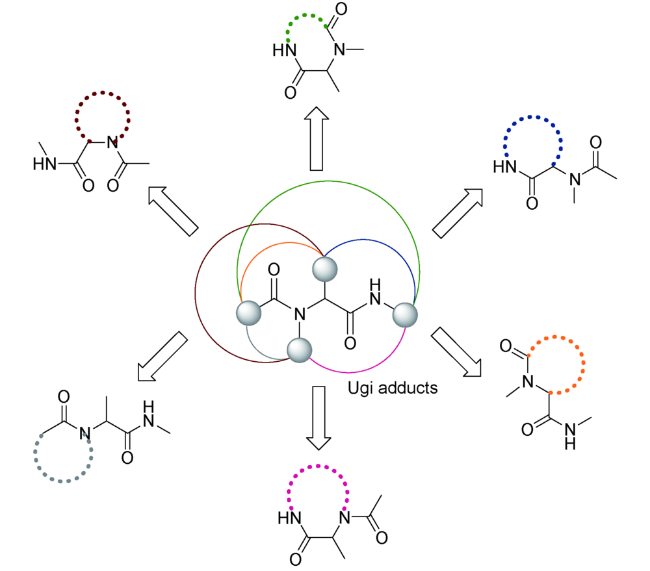
该法与传统方法相比,避免了多步纯化操作以及保护、脱保护等繁琐过程,具有极高的步骤和原子经济性,一揽子解决了当今合成化学面临的待合成分子数量庞大、结构复杂等诸多难题,其优势不言而喻。
目前,Ugi串联反应在杂环化物合成领域方兴未艾,通过Ugi反应与后续反应(post reaction)[26, 27]或多米诺过程(domino process)[28, 29]联用,实现目标化合物库的快速高效构建,涌现出诸如:Ugi/Heck[30]、Ugi/Michael[31]、Ugi/Wittig[32]、Ugi/Joullie[33]、Ugi/Buchwald-Hartwig[34]等优秀的串联反应策略,成功合成多种杂环骨架,如:喹啉、异喹啉、吲哚、苯并呋喃、苯并噻唑、异口恶唑等。Ugi/Diels-Alder(UDA)串联反应集Ugi与Diels-Alder反应优势于一身,在构建杂环化合物方面展现出了巨大优势和无穷潜能,受到越来越多有机化学工作者的青睐。这一领域的研究成果层出不穷,形成了一套独有的设计思路与理论体系。本文将以不同类型的DA反应为切入口,对UDA串联反应的研究进展作一综述。
2 呋喃作为双烯体的UDA串联反应
呋喃是一类富电的芳环,可作为加成DA反应的天然双烯体,参与分子内Diels-Alder反应(Intramolecular Diels-Alder reaction,IMDA)。以呋喃为双烯体的UDA串联反应构建杂环化合物的一般设计思路为:把呋喃和与之加成的亲双烯体引入Ugi产物分子中,通过双官能团间的反应,引发后续的IMDA环加成。
(1)烯作为亲双烯体
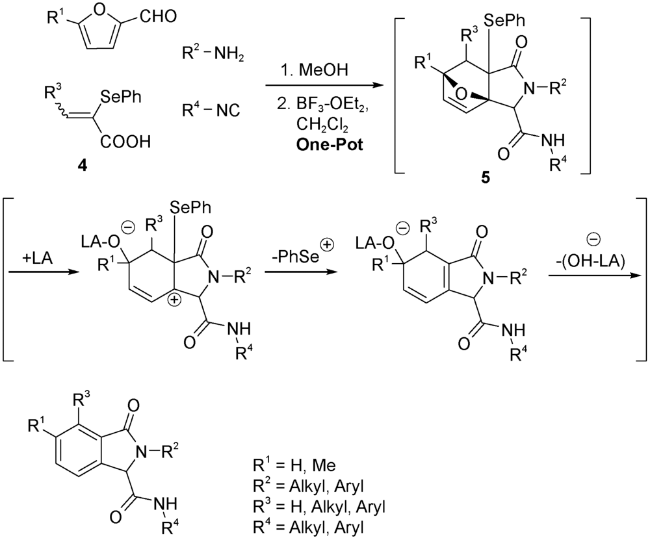
Huang等[37]报道了一种利用UDA串联反应,“一锅煮”合成异吲哚啉类化合物的方法。首先让糠醛、胺、2-苯硒基烯酸4和异腈在甲醇中反应。待反应生成三环中间体5完全后,换用CH2Cl2作溶剂,用路易斯酸——三氟化硼(BF3)断开氧桥,通过羟基和苯硒的消除,推动芳构化实现异吲哚啉类化合物的合成(图式3)。
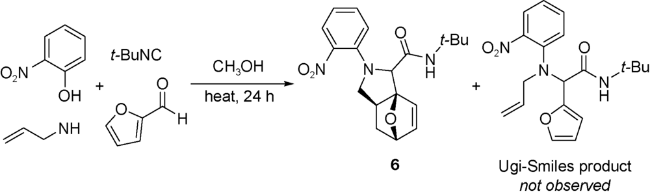
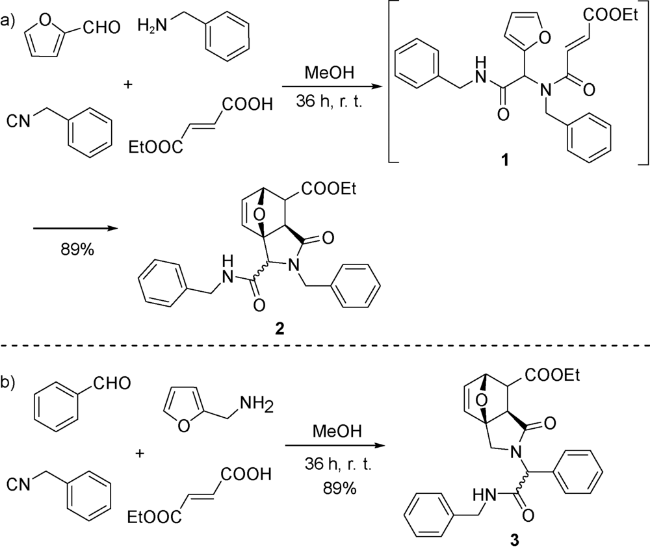
Paulvannan[35]首次应用固相合成法实现UDA串联策略。该策略使用酸不稳定树脂作为固相,将和固相连接的胺7与过量的羧酸、醛和异腈在甲醇与二氯甲烷的混合溶剂中反应36 h后,加入三氟乙酸(TFA)使产物与固相分离,得到化合物8(图式5)。
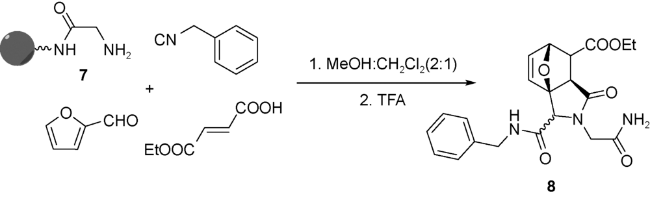
Schreiber等[40]以高容量聚苯乙烯树脂作为固相,将N-保护苄胺9连接于载体之上,通过UDA串联反应生成三环化合物10。他们通过X射线单晶衍射确定了产物10的主要构型为顺式。10再经过三步反应,可以得到结构更为复杂的天然多环化合物类似物11(图式6)。
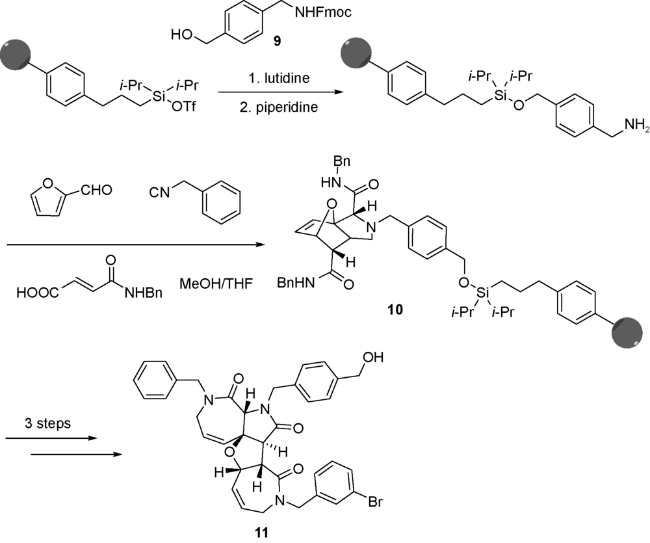
Oikawa等[41]将5-羟甲基-2-呋喃甲醛12在氯化碘(ICl)的作用下,固化在聚乙二醇端基改造材料上,得到连接产物;该步也可使用三氟甲磺酸甲酯催化,如使用后者,反应时间由原来的1 h延长至37 h,收率也有所降低(图式7)。Oikawa等选择这种固相连接方式的目的有以下两点:(1)在UDA串联反应中,糠醛较其他三个组分,具有不可替代性,因此选择糠醛与固相连接;(2)该方式有助于分离出连接聚合物的亚胺中间体,这就相当于一个反应池,通过加入不同的酸和异腈,得到不同的UDA产物。将固化后的糠醛13与过量的胺、苄基异腈和富马酸单酰胺反应,用三氟甲磺酸使产物与固相分离,即得到14。实验中还观察到去羟甲基副产物15生成,并且随着温度和底物浓度的升高,副产物的比例增大。
(2)炔作为亲双烯体
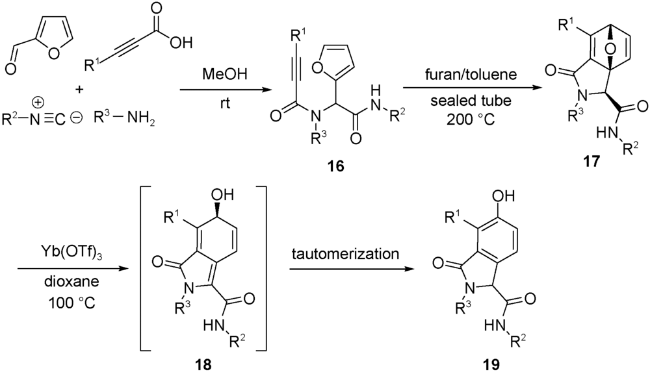
Wright等[42]首次报道了一种使用炔酸代替烯酸作为亲双烯体的UDA串联反应,制备出一系列异吲哚啉衍生物。得到Ugi产物16不需要任何催化,直接加热至200 ℃就能触发IMDA反应,完全转化为成环产物17。反应表现出良好的立体选择性,主要构型的异构体在产物中的比例大于90%,易于分离纯化。通过X射线单晶衍射,确定了17中主要异构体为顺式构型,这与Schreiber等[40]报道的烯基加成的UDA串联反应产物的主要构型恰好相反。反应所使用催化氧桥断裂的路易斯酸为三氟甲磺酸镱(Yb(OTf)3)。氧桥断裂后形成的中间态18,经过异构重排后,转化为6-羟基异吲哚酮类化合物19(图式8)。McCluskey等[43]在Wright等的研究基础上,讨论了该类反应的底物适用性。对于异腈组分,无论是芳香异腈还是脂肪族异腈均适用于该类反应;对于胺组分,缺电子和富电子型苯胺以及脂肪族胺都表现出良好的底物适用性。
3 吡咯作为双烯体的UDA串联反应
除了呋喃,吡咯也能做作为亲双烯体参与的UDA串联反应。但是目前尚无关于单纯吡咯作为UDA串联反应双烯体的报道,UDA串联反应使用的是一类N-磺酰化吡咯。
(1)烯作为亲双烯体
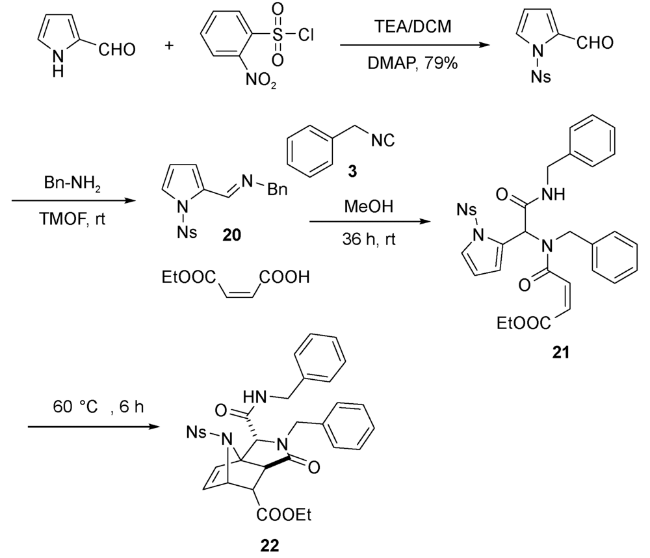
Paulvannan[44]用N-保护的吡咯亚胺20,与苄基异腈和马来酸单乙酯在甲醇中反应,生成Ugi产物21,进而引发后续的IMDA反应,得到与呋喃产物结构相似的三环内酰胺衍生物22(图式9)。如用吡咯甲醛和苄胺直接参与反应收率较差,这是因为它们形成亚胺的能力较弱。如用亚胺20参与反应,收率则明显提高。用富马酸单乙酯代替马来酸单乙酯参与反应,随即生成环化产物22,不需要额外加热。这说明烯酸的反式构型更有利于IMDA加成进行。但无论使用顺式还是反式丁烯二酸单酯,产物均为单一构型。
(2)炔作为亲双烯体
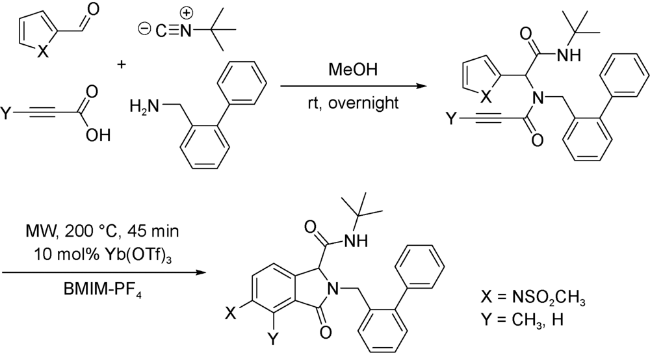
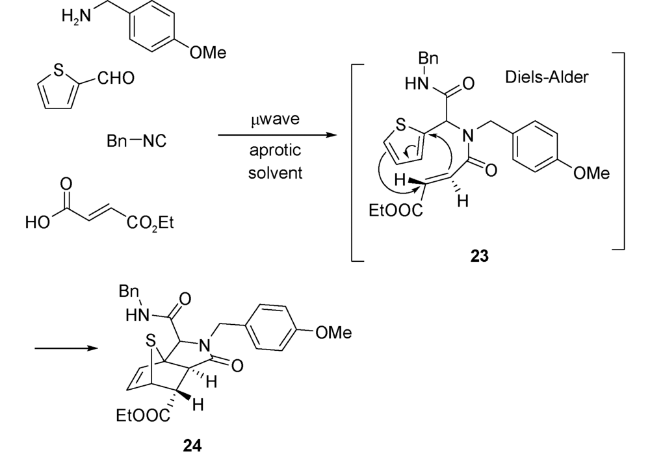
4 以噻吩为双烯体的UDA串联反应
5 口恶唑作为双烯体的UDA串联反应
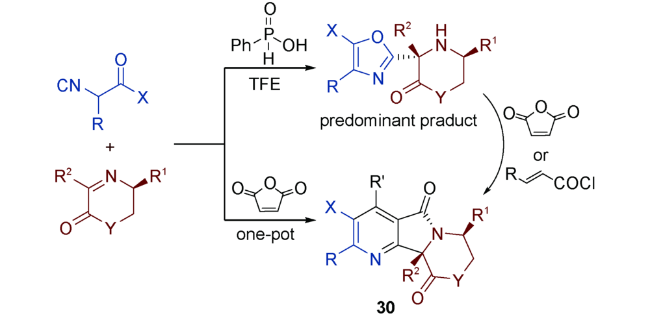
口恶唑同其他五元富电芳环一样,也能作为双烯体参与DA反应。但鲜有关于口恶唑直接作为取代基参与Ugi反应的报道[47],这类反应中口恶唑引入反应体系往往是通过原位生成的。
这些基于异腈三组分反应合成的取代口恶唑在结构中都含有一个游离的仲氨基,相当于预留了Ugi反应位点。通过加入α,β-不饱和酰卤或酸酐,酰化氨基的同时引入亲双烯体,对分子内的口恶唑进行DA加成。这就是以口恶唑为双烯体的UDA串联反应的主要合成策略。需要注意的是,上述四组分反应并非严格意义上的Ugi反应,但是反应历程和产物结构又与经典的Ugi反应极为相近,不少学者将其称之为Ugi型(Ugi-type)反应[52,53,54]。因此,我们将这类四组分反应并入广义Ugi反应的范畴并加以讨论。以口恶唑为双烯体的UDA串联反应策略也为杂环化合物的合成提供了新的思路——双烯体既可以由反应底物直接引入,也可以通过反应原位生成。
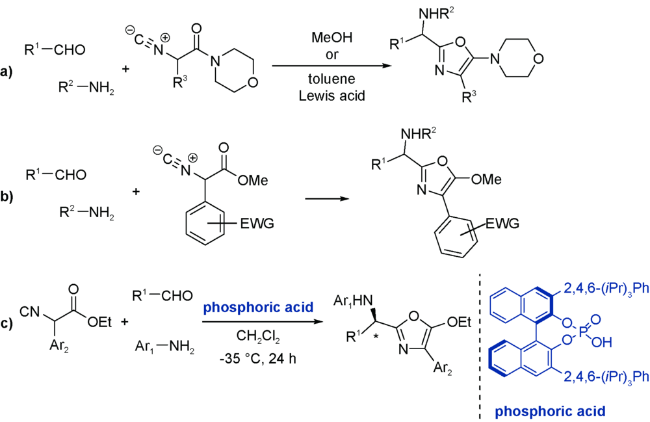
(1)烯作为亲双烯体
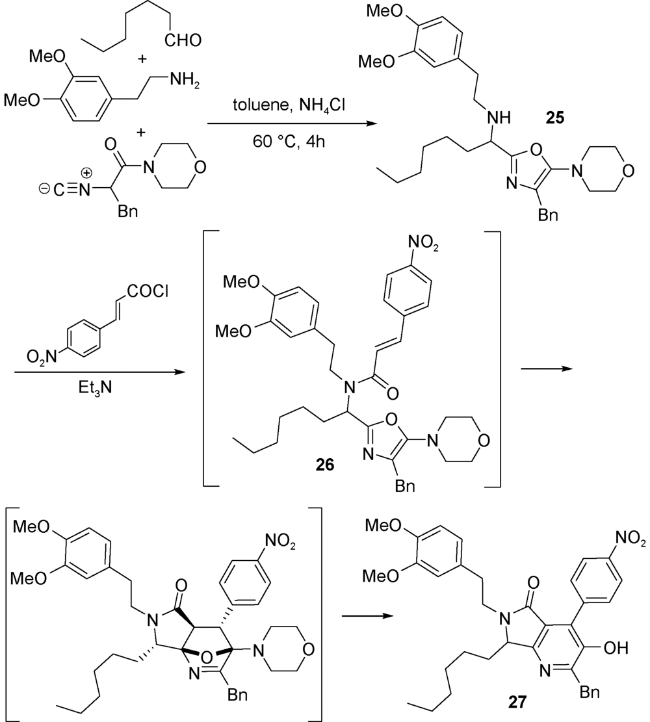
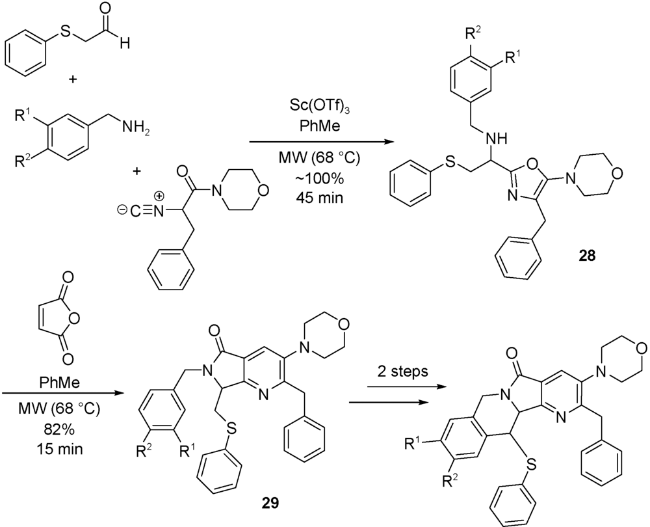
Islas-Jácome等[53]通过醛、苄胺和异腈在三氟甲基磺酸钪(Sc(OTf)3)的催化下,通过微波辅助加热,得到取代口恶唑28,然后在反应体系中加入酰化试剂顺丁烯二酸酐,经过一系列分子内反应,生成UDA串联反应产物——吡咯并[3,4-b]吡啶-5-酮29。29再经过S-氧化和Pummerer两步反应后,最终生成一系列多环化合物(图式14)。
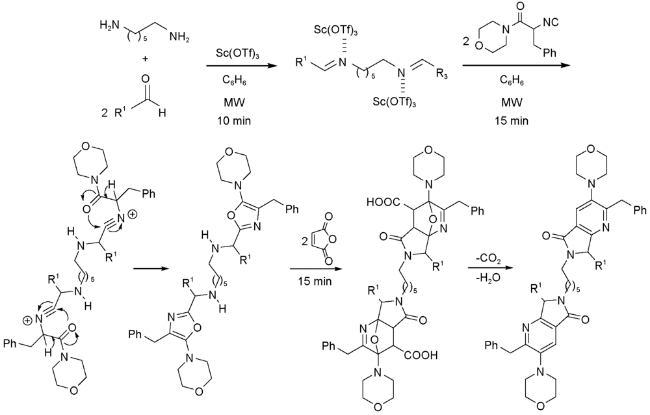
González-Zamora等[56]利用UDA串联反应,“一锅煮”合成了具有广泛的生物活性的六亚甲基酰胺(Hexamethylene-bisacetamide,HMBA)类似物。反应创新点在于应用二元胺(1,6-己二胺)作为底物,在两端同时构建吡咯并[3,4-b]吡啶-5-酮环系(图式15)。
Chen等[54]以环状亚胺为底物与异腈反应,三氟乙醇作溶剂,在苯基亚磷酸的催化下,立体选择性合成手性口恶唑。与顺丁烯二酸或丙烯酰氯反应后生成三环化合物。也可以通过异腈、亚胺和顺丁烯二酸酐“一锅煮”,合成三环化合物(图式16)。对反应历程的研究表明:无论是“两步走”,还是“一锅煮”,反应都要先生成口恶唑环再进行IMDA加成。
(2)炔作为亲双烯体
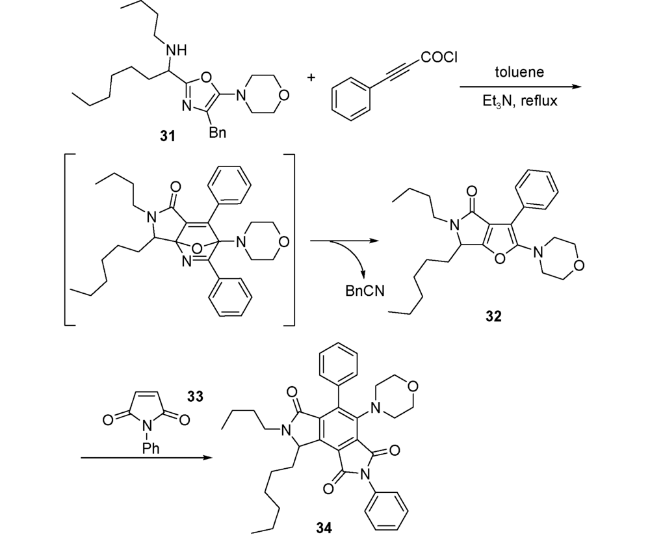
Zhu等[57]用合成口恶唑31与苯基丙炔酰氯在甲苯中反应,随着苄腈的离去,生成了吡咯并呋喃中间体32。然后在反应体系中再次加入环状亲双烯体吡咯二酮33,继续进攻32分子中新生成的呋喃环,继而生成六取代苯34。反应历经了IMDA/retro-DA/DA进程(图式17)。
6 1,2,4-三嗪作为双烯体的UDA串联反应
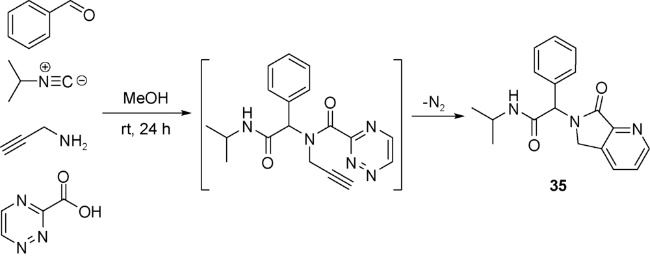
Akritopoulou-Zanze等[58]报道了一种Ugi反应与反电子需求的Diels-Alder反应(inverse electron demand Diels-Alder reaction)的串联策略。反应突出特点在于使用缺电杂环1,2,4-三嗪作为双烯体。1,2,4-三嗪甲酸表现出良好的Ugi反应性,在与苯甲醛、异丙基异腈和炔丙胺“一锅煮”的反应中,以32%的收率得到吡咯并[3,4-b]吡啶-7-酮35, 推测反应的推动力来自N2的释放(图式18)。
7 苯作为双烯体的UDA串联反应
8 不饱和键和芳环共同作为双烯体的UDA串联反应
Yang等[60]应用UDA串联反应首次合成出一系列苯并呋喃、吲哚和苯并噻吩等杂环化合物。如图式20所示,Ugi产物分子37中,呋喃和其上共轭双键作为双烯体共同参与后续的IMDA反应。反应得到Ugi产物38,在氧气氛围中,让其与活性炭在甲苯中回流,氧化芳构化生成三环化合物39。反应也可用2,3-二氯-5,6-二氰基-1,4-苯醌(DDQ)代替活性炭。同时,N-甲基吡咯、噻吩甚至是苯环都可以代替呋喃参与该类反应,表现出良好的底物适用性。
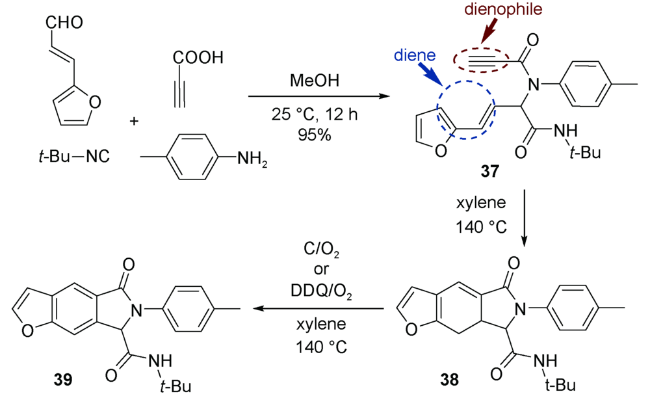
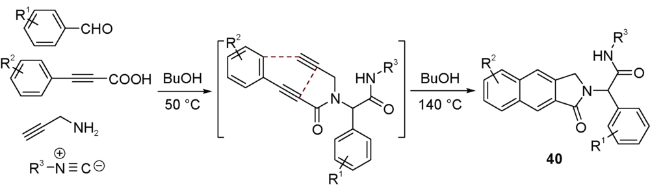
上述反应使用烯烃作为亲双烯体,如果使用炔烃参与反应是否可以避免使用分步反应实现氧化芳构化。2014年,Eycken等[61]就开发了一种炔烃参与的无金属、“一锅煮”UDA串联反应构建苯并[e]异吲哚40的新方法(图式21)。该反应还可以通过溶剂调控封闭环路径的选择性,实现苯并[f]异吲哚的多样性导向合成。
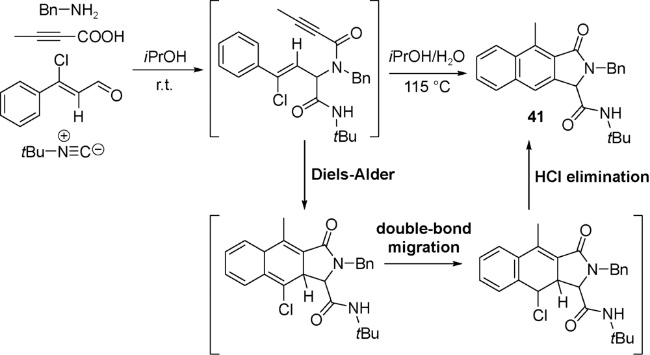
9 Ugi/retro Diels-Alder串联反应
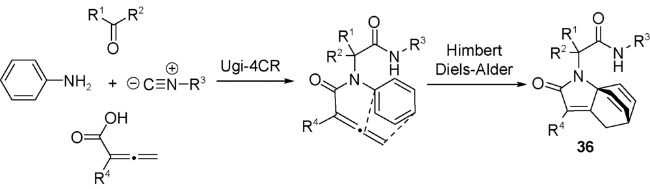
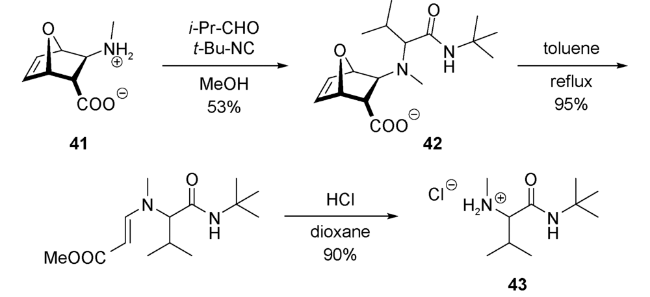
2009年,Basso等[63]利用不饱和β-氨基酸衍生物与异丙基醛和叔丁基异腈发生分子内Ugi反应,得到42。将42在甲苯中回流,引发逆Diels-Alder反应(retro Diels-Alder reaction)。随着呋喃离去,烯胺的水解,得到一系列氨基酸衍生物43(图式23)。41的分子中含有的刚性桥环骨架是很好的手性助剂,使分子内Ugi反应表现出良好的立体选择性。该Ugi/retro Diels-Alder串联反应在手性氨基酸衍生物的合成中具有广阔的应用前景。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
10 结论
尽管如此,Ugi/Michael串联反应构建小分子杂环化合物仍存在以下不足之处:(1)市售异腈的种类较少,影响了酰胺上取代基的多样性。(2)UDA串联反应双烯体一般都位于Ugi反应的醛和胺组分上,而亲双烯体一般为炔酸或烯酸。这样的组合既方便引入反应位点,又具有良好的Ugi反应性。但是过于依赖这样底物组合会造成产物骨架雷同,影响产物分子结构的多样性。目前尚无从异腈引入双烯体和亲双烯体的文献报道,今后对这方面的研究可能会成为该领域的开拓方向。(3)UDA串联反应严重滞后于DA反应的发展,应当从DA反应中寻找灵感,将最新的研究成果应用于UDA串联反应中去,设计新的双烯体和亲双烯体引入反应体系,为UDA串联反应注入新的生机与活力。(4)UDA产物分子中往往含有桥环(氧桥、氮桥、硫桥等),需使用催化/开环/芳构化的策略,使其转化为平面结构,以利于后期的生物活性筛选和构效关系的说明。
尽管UDA串联反应还存在着诸多不足,但是其在构建杂环化合物方面有得天独厚的优势以及在多组分化学中有独特魅力,相信通过有机化学工作者的不断努力,它的明天一定会更美好。也希望本文能够为有机合成及药物化学工作者提供一定的参考。