




Contents
1 引言
2 生物材料表面修饰相关的光化学反应
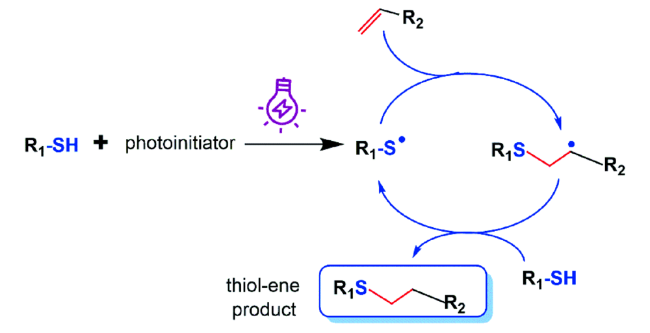
2.1 硫醇-烯烃/炔烃光点击反应
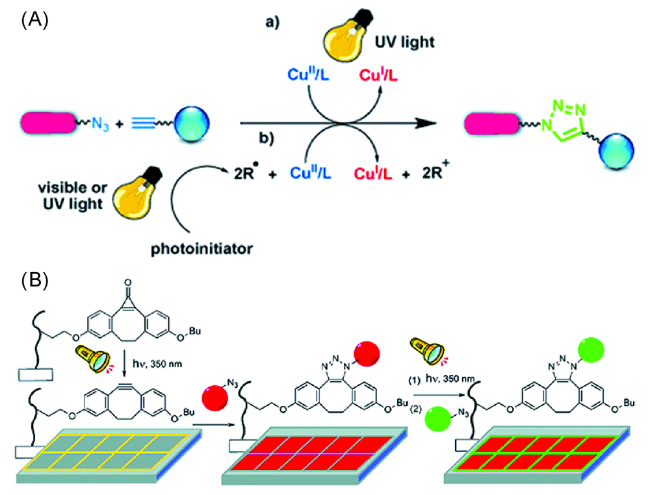
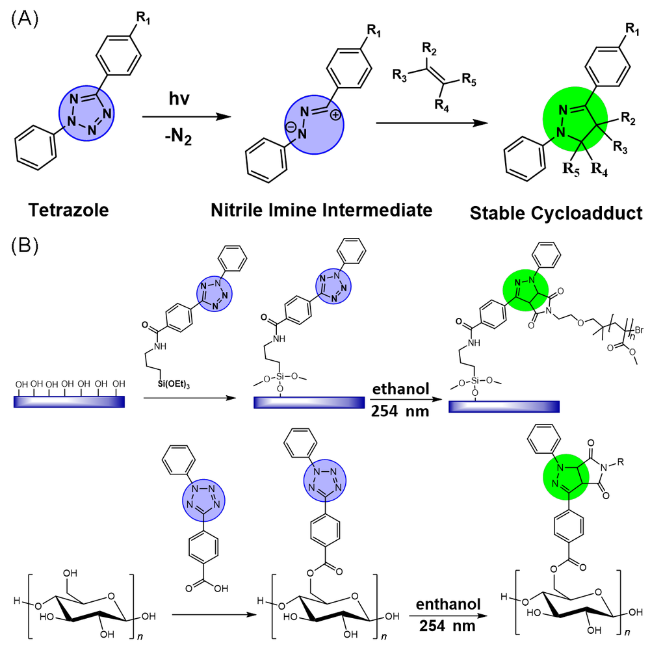
2.2 光引发的环加成反应
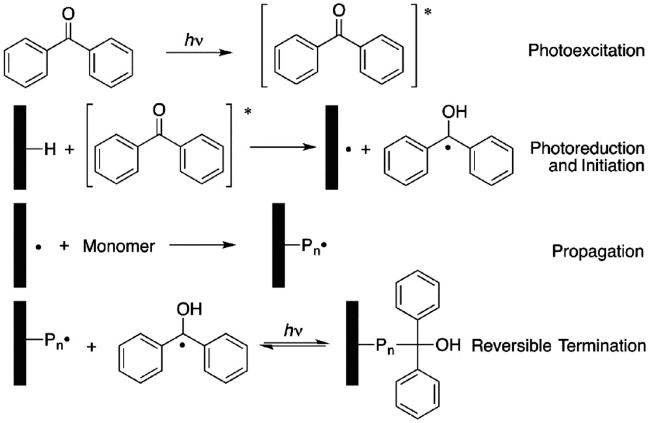
2.3 表面光接枝聚合反应
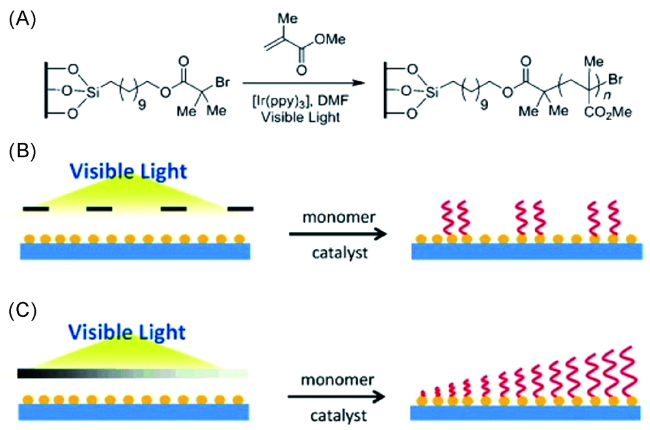
2.4 光不稳定基团反应

2.5 苯基叠氮化合物反应

3 表面光化学反应制备生物医用材料的研究
3.1 化学仿生表面
3.2 薄膜凝胶的硬度调控
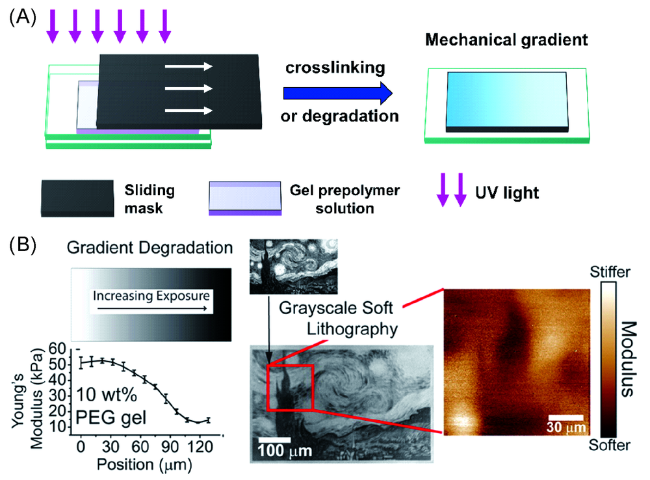
3.3 图案化和梯度表面
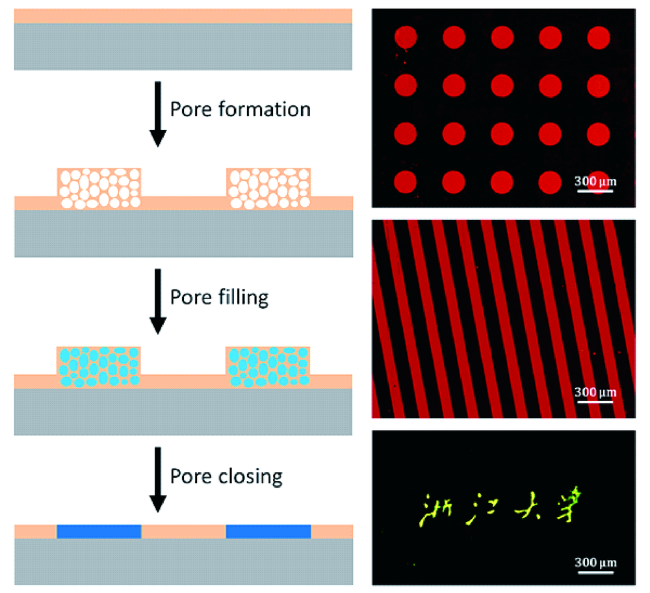
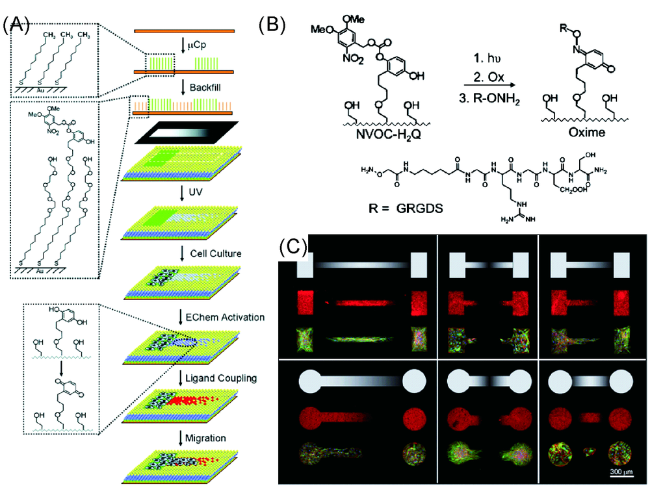
图10 (A)制备动态图案和梯度化表面的流程;(B)电化学和光化学方法共同在表面固定配体[110]; (C)细胞在RGD梯度表面的黏附和迁移图片[111]Fig. 10 (A) Flow diagram for preparing dynamic patterns and gradient surfaces;(B)Combined electroactive and photochemical strategy for chemoselective immobilization of ligands[110];(C) Representative images for cell attachment and migration on RGD defined gradients[111] |
3.4 光响应动态生物材料表面

3.5 微液滴阵列表面

图12 微液滴阵列细胞筛选平台[132]。(A) 不同尺寸大小的超亲水和超疏水区域设计;(B)微液滴阵列照片;(C)微液滴阵列用于细胞筛选的工作流程图(比例尺1 mm)Fig. 12 Droplet-microarray(DMA) cell screening platform[132].(A) Different sizes of the super-hydrophilic spots and super-hydrophobic borders;(B) Photographs of droplet microarrays;(C)Schematic diagram of cell screening using a DMA platform(scale=1 mm) |
3.6 生物材料高通量制备平台

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
图13 (A)硬度-配体浓度正交高通量凝胶制备原理图;(B)正交梯度凝胶表面U373-MG细胞miR18a表达情况分布;(C)巨噬细胞分泌的外源可溶性因子对U373-MG细胞miR18a表达影响[144]Fig. 13 (A)Preparation schematic of matrix stiffness and fibronectin density orthogonal high-throughput gel;(B) The distribution of miR18a expression in U373-MG cells on orthogonal gradient gel;(C) The effect of exogenous macrophage-derived soluble factors on ECM-sensitive regulation of miR18a[144] |