



Contents
1 Introduction
2 Potassium-ion battery cathode
2.1 Prussian blue analoguescathode
2.2 Layered metal oxidescathode
2.3 Polyanioniccathode
2.4 Organic compound cathode
3 Potassium-ion battery anode
3.1 Intercalation anode
3.2 Alloying anode
3.3 Conversion anode
4 Electrolyte
5 Conclusion
1 引言
当今社会科技与经济的迅猛发展使得全球环境污染和能源的问题日益突出。可充电锂离子电池已经广泛作为能源储存中介应用于便携式电子产品。然而, 锂资源的稀缺以及随之而来的锂离子电池成本的提高, 很大程度上限制了其在大规模储能中的应用, 因此在能源储存体系中寻求无资源限制、高能量密度的二次电池体系成为亟待解决的问题。钾和锂同属第一主族元素, 具有相似的物理化学性质。钾资源在地球的丰度较高(2.09 wt%)、分布广泛、易于获取, 且与锂元素不同, 钾元素不会和铝集流体发生合金化反应, 使得负极可以用价格低廉的铝箔作为集流体, 从而大大降低了钾离子电池体系的成本。有趣的是, 钾的标准电极电势是-2.93 V, 比较接近于锂的-3.04 V, 使得钾离子电池具有较高的工作电压和能量密度。碱金属离子当中钾离子的路易斯酸性最弱, 这使得钾离子在电解液中以及电极与电解液表面有很大的迁移数和迁移率;在电解液(溶剂化离子)中钾离子的斯托克半径较小, 所以钾离子电池中K+的化学扩散系数高于Li+在锂离子电池中的化学扩散系数。
与锂离子电池相比, 钾离子电池与其构造及工作原理相似:钾离子在正负电极之间可逆的嵌脱引起电极电势的变化而实现电池的充放电, 是典型的“摇椅式”储能机理。电池的正负极分别由两种不同的能够可逆嵌脱钾离子的材料构成。充电时, 钾离子从正极脱出, 进入电解质中, 且通过外在电场力的作用迁移到负极;同时电子通过导通的外电路由正极流向负极, 从而保证正负极的电荷平衡。放电过程则与之相反。钾离子电池的充放电原理示意图如图1所示。
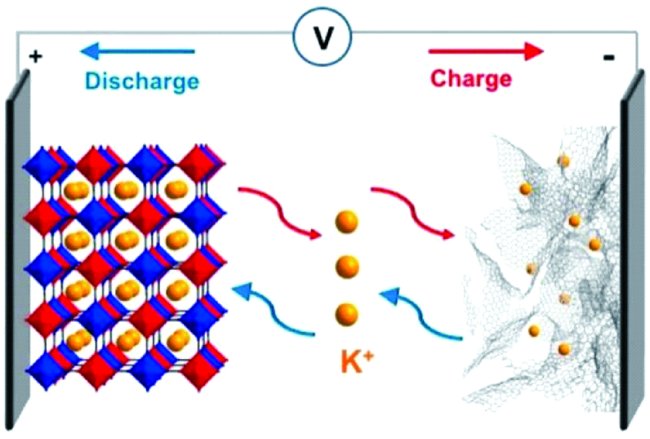
虽然这些鲜明的优势都使得钾离子电池在大规模储能系统中展现出光明的应用前景, 然而钾元素具有更高的相对原子质量, 这在理论上也限制了钾离子电池的能量密度。同时, 钾离子半径较大[2], 其本身嵌脱动力学过程较慢, 并且电极材料在反复嵌脱过程中承受较大的结构应力, 最终导致钾离子电池电化学循环性能和倍率性能不尽人意。因此, 开发有利于钾离子可逆嵌脱的电极材料和与之相匹配的电解液是提高钾离子电池电化学性能的关键。目前, 国际上众多科研工作者对钾离子电池开展研究课题, 得到诸多令人欣慰的科研成果。本文主要围绕正极材料、负极材料以及电解液三方面的研究工作, 对钾离子电池的研究近况进行介绍, 同时对钾离子电池未来发展方向做出展望。
2 钾离子电池正极材料
2.1 普鲁士蓝类正极材料
普鲁士蓝及其类似物的刚性开放框架结构具有大的间隙, 为离子半径较大的钾离子的可逆嵌脱提供了丰富的活性位点和传输通道。普鲁士蓝类化合物的通式为KxM[M'(CN)6]1-y·□y·mH2O(M是Fe、Co、Mn、Ni、Cu、Zn等或其中的几种组合;M'一般是Fe;□是氰基空位;0≤x≤2, y<1), 其结构为面心立方结构, 具体的晶体结构可用图2来表示。普鲁士蓝类正极材料具有钙钛矿结构, 晶格中过渡金属M与亚铁氰根按照Fe-C≡N-M排列形成三维骨架结构, Fe离子与M离子按照立方体状排列, C≡N位于立方体的棱上。当掺杂的过渡金属元素M(Fe、Mn)是具有电化学活性的, 其晶格中可以包含两个不同的氧化还原活性位点(Mn/Mn+1和Fe2+/Fe3+), 理论上可实现约155 mAh/g的容量, 然而与此同时, 两电子转移的特性也使得该材料的结构不稳定, 易发生不可逆的转变;当掺杂的过渡金属M(Ni、Cu、Zn等)是电化学惰性的, 该类材料只能实现一个电子的转移, 但也正是因为这些过渡金属元素的电化学惰性使得它们起到支撑开放结构框架的作用, 所以含有此类过渡金属元素的材料表现出了良好的循环性能和优异的库仑效率。材料成本低廉、制备简单、环境友好等特性使得普鲁士蓝类材料在大规模储能方面具有广阔的应用前景。
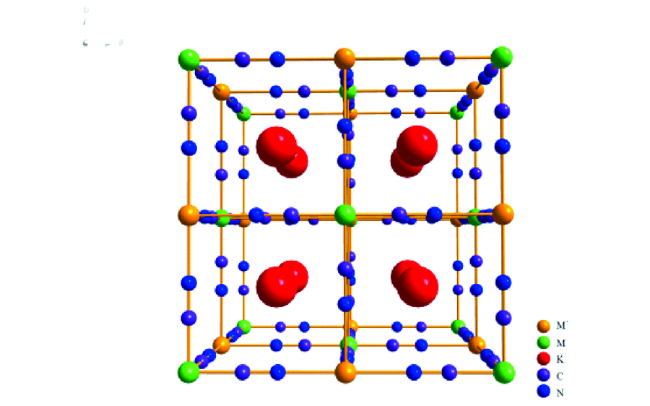
人们认为在正负极之间可逆嵌脱的钾离子数量会影响电池的电化学性能和材料的晶型结构, 增加正极材料中K+的含量可有效提升全电池能量密度和可逆比容量。Chen等[5]采取添加KCl来增大反应介质中K+浓度, 获得了富钾态普鲁士蓝类化合物K1.92Fe[Fe(CN)6]0.94·0.5H2O, 在电解质体系KClO4/PC中具有133 mAh/g的高比容量和良好的长循环稳定性, 0.1 C电流密度下循环200圈后容量保持率为92.8%。Goodenough等[6]将制备出的K1.89Mn[Fe(CN)6]0.92·0.75H2O 作为钾离子电池正极材料时, 可以得到142 mAh/g的放电比容量。由于与N相配位的高自旋Mn3+/Mn2+和与C相配位的低自旋Fe3+/Fe2+具有接近的能势, 该材料在放电过程中展现了两个电压集中在3.6 V的放电平台。当使用亚铁氰化钾和亚铁氰化钠分别与锰的硝酸盐发生共沉淀反应时, 前者的产物颗粒尺寸比后者的小, 表明亚铁氰化锰钾的结晶速率比亚铁氰化锰钠的快。于是他们在反应体系中同时加入KCl和NaCl, 发现Na+会诱导颗粒尺寸较小的初级粒子聚集形成直径约350 nm的二级粒子, 该策略有效地改善了材料的循环性能。
研究者发现当框架中的M采用不同的过渡金属时, 可以获得丰富的结构体系, 表现出不同的储钾比容量和倍率性能。Ji等[7]合成一系列普鲁士白类似物KxMFe(CN)6·mH2O(M=Fe、Co、Ni和Cu), 其中FeFe-PW(PW:普鲁士白)具有两电子转移特性, 在20 mA/g的电流密度下可以达到110 mAh/g的可逆容量;而NiFe-PW、CoFe-PW和CuFe-PW表现出单电子反应机制。CoFe-PW作为钾离子电池正极材料时只有约60 mAh/g的低放电容量, 而钠类似物Na2CoFe(CN)6却表现出两电子转移特性, 他们推测这种差异可能是因为K+和CN—之间的强键合作用导致电子云密度向K+偏移, 产生的诱导效应使得Co3+/Co2+的氧化还原电位增加, Co2+在充电上限电压的限制下不能被氧化[8]。
Komaba等[9]将通过水溶液沉淀法制得的K1.75Mn[Fe(CN)6]0.93·0.16H2O和K1.64Fe[Fe(CN)6]0.89·0.15H2O作为钾离子电池正极材料, 前者具有3.8 V的放电电压平台和137 mAh/g的放电容量, 后者则表现出3.4 V的放电电压平台和130 mAh/g的放电容量。根据钠电上Na2Fe[Fe(CN)6]正极材料的研究[10], 他们指出两者容量的差异归因于[Fe(CN)6]结构缺陷的数量不同, 这些结构缺陷会减少电化学活性中心, 使得容量利用率降低。此外, 他们还将石墨作为钾离子电池负极材料与K1.75Mn[Fe(CN)6]0.93·0.16H2O正极材料组装成全电池, 表现出110 mAh/g的可逆比容量, 且在3.8 V的放电平台下具有536 Wh/kg的能量密度, 与锂电中LiCoO2表现出的532 Wh/kg能量密度相当。他们还提出该正极材料在钾离子的嵌入和脱出过程中经历了单斜晶相⇌立方相⇌四方晶相的可逆相变。其中, 第一个两相变化归因于处于旋转态的MnN6和FeC6八面体转变为未旋转的规整状态, 第二个两相变化是由高自旋Mn3+的姜-泰勒效应引起的。
Liu等[11]通过水热法制备出KFeⅡ[FeⅢ(CN)6]纳米颗粒, 其作为正极材料时具有超长循环寿命, 即在电流密度100 mA/g下循环1000周后容量保持率为80.49%且电压平台在循环过程中没有明显的变化。他们指出钾离子的嵌入与脱出是一个温和的固溶过程, 还通过非原位XRD和非原位傅里叶转换红外光谱对钾离子的嵌脱机制进行了分析, 发现在充放电的过程中水分子始终存在, 水分子虽然对容量并没有贡献, 但却可以起到支撑框架结构的作用。利用常规水溶液共沉淀法制备的普鲁士蓝类化合物, 其沉淀溶解平衡常数极小, 导致晶体的成核和生长在自发沉淀期间通常是一个快速过程, 造成产物差的结晶度和颗粒的严重聚集。研究表明材料的晶粒尺寸对其电化学性能有很大的影响, 柠檬酸盐等络合剂可以与过渡金属离子M2+发生螯合作用从而减缓产物结晶速率。Nazar等[12]通过柠檬酸盐辅助络合沉淀法合成了不同尺寸的普鲁士白类似物K1.7Fe[Fe(CN)6]0.9。当晶粒尺寸为20 nm时, 拥有接近140 mAh/g的理论容量和两个明显的放电电压平台分别为4.0 V和3.2 V;通过调控柠檬酸盐的量使晶粒尺寸增大时, 其电化学性能下降。
众多研究者发现通过添加柠檬酸盐或氯化钾等方式辅助沉淀普鲁士蓝类似物, 减少了传统普鲁士蓝类材料结构缺陷多、结晶水含量大等突出问题, 进一步提高了材料稳定性, 增加了初始充放电容量。然而普鲁士蓝类材料依然面临着高倍率下性能匮乏、长循环下容量衰减快等诸多挑战。因此, 制备出低缺陷、含水少的普鲁士蓝类材料是显著提高该类材料电化学性能表现的关键。
2.2 层状过渡金属氧化物类正极材料
层状过渡金属氧化物由于其高理论能量密度、良好的结构稳定性、低廉的制备成本以及环境友好的特点, 在锂、钠二次电池电极材料中得到广泛应用, 因而也成为钾离子电池正极材料的合理选择。层状过渡金属氧化物的通式为KxMO2(M是Fe、Co、Mn、V等或其中的几种组合)。根据钾离子在层状过渡金属层间排列方式的不同, 钾基层状氧化物可分为:O3型(ABCABC堆叠)、P2型(ABBA堆叠)和P3型(ABBCCA堆叠)三类。其中, P、O表示不同密堆积方式中钾离子处在不同的配位环境(P为棱形、O为八面体);2、3表示过渡金属离子占据不同位置的数目, 是由氧离子堆积方式决定的。
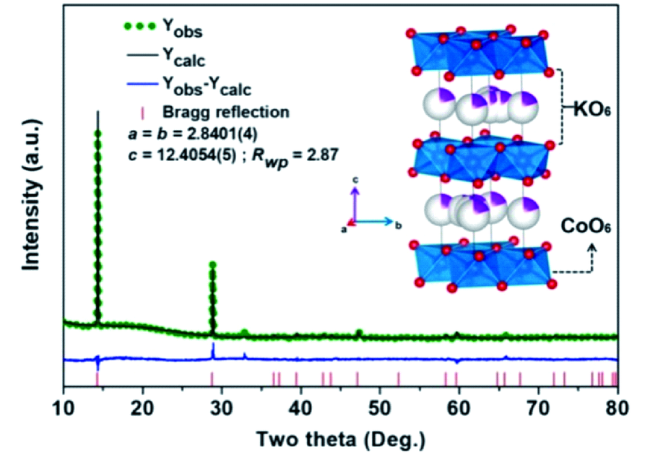
Komaba等[15]将P2型和P3型的KxCoO2作为钾离子电池正极材料, 在2.0~3.9 V的电压范围内有多个电压平台出现, 可以提供60 mAh/g的可逆容量, 且具有良好的循环稳定性和倍率性能。他们通过研究O2-LixCoO2、P2-NaxCoO2和 P2-KxCoO2分别作为锂电、钠电和钾电正极材料, 发现可充电碱金属电池的工作电压与CoO2的层间距有关。Ceder等[16]通过固态法合成了具有层状结构的P2-K0.6CoO2材料, 将其作为钾离子电池正极材料时K+的可逆嵌脱区间为0.33~0.68, 表现出80 mAh/g的可逆容量。在电流密度100 mA/g下循环120周后, 容量保持率为60%, 且在每个倍率下都表现出优异的循环稳定性。为了解释正极材料嵌脱钾的机理, 研究人员利用原位XRD技术阐明了材料是通过高度可逆的拓扑定向反应来储存K+的。Wang等[17]利用两步自模板法制备出由初级纳米片聚集的P2-K0.6CoO2微球, 其精修X射线衍射谱图及结构示意图如图3所示。得益于独特的分级结构, 材料具有较高的比容量和出色的循环性能。在10 mA/g电流密度下比容量为82mAh/g, 在40 mA/g电流密度下循环300圈后容量仍有65 mAh/g, 每圈只有0.04%的容量衰减。
近年来锰钾氧化物因锰资源储量丰富, 而且锰价格低廉、无毒, 而成为电极材料中的研究热点。Vaalma等[18]报道了将具有层状结构的水钠锰矿K0.3MnO2用于钾离子正极材料, 在1.5~3.5 V的电压区间内具有良好的循环稳定性, 可逆容量约为70 mAh/g, 但当上限截止电压提高到4.0 V时, 结构稳定性变差, 导致容量的不可逆衰减。Ceder等[19]研究了不同放电电位窗下P3-K0.5MnO3的电化学性能:在电压范围为1.5~3.9 V的CV曲线中, 存在对应的氧化还原峰;当上限电压增加为4.2 V时的CV曲线中, 在 3.7 V和4.1 V的位置出现氧化峰, 却没有相应的还原峰。随后, 他们对材料进行原位XRD测试, 结果证明其结构在高电压范围内发生了P3到O3的不可逆相变, 这与低电压范围内P3⇌O3⇌X的可逆转变不同。向锰氧化合物中掺杂Ni、Co、Fe等过渡金属元素可以有效抑制复杂的相变, 增强结构的稳定性能, 获得更好的电化学可逆性。Mai等[20]通过静电纺丝技术得到了K0.7Fe0.5Mn0.5O2纳米线, 原位XRD结果显示该材料在钾离子嵌脱的整个过程中始终保持稳定的层状框架结构。由于具有快速的K+扩散通道和三维电子传导网络, K0.7Fe0.5Mn0.5O2作为正极材料与软碳负极材料组装成钾离子全电池时, 在20 mA/g的电流密度下可以获得119 mAh/g的容量, 循环250周后容量保持率为76%。此项研究工作推动了由地球丰富元素组成的高性能钾离子正极材料的广泛研究。最近, Wang等[21]通过改进的溶剂热法合成出由初级纳米粒子自组装而成的均匀K0.65Fe0.5Mn0.5O2(P2-KFMO)微球。作为钾离子正极材料时, 在20 mA/g电流密度下具有高度可逆的储钾容量(151 mAh/g), 在100 mA/g电流密度下循环350周后, 表现出78%的容量保持率。他们指出活性材料良好的结构稳定性和优异的电化学性能归因于其独特的分层结构。微粒级的二级K0.65Fe0.5Mn0.5O2微球减小了P2-KFMO与电解质的接触面积, 有效降低了两者之间的副反应;同时纳米级的初级粒子能够缩短K+扩散距离, 提供稳定的K+和电子传输路径。此外, P2-KFMO微球中的孔隙可以容纳充放电过程中电极的体积变化, 使微球表面上生成的钝化层更加稳定, 从而实现了材料的长期循环稳定性。
Li等[22]利用水热法合成了网状V2O5·0.6H2O干凝胶, 通过将结晶水限制在V2O5层中, 可以扩大材料的层间距, 从而获得优异的钾储存能力。首圈放电比容量可达224.4 mAh/g, 在50 mA/g的电流密度下循环500周后能够提供103.5 mAh/g的放电容量。Zhu等[23]同样采用水热法制备出了层状结构的K0.5V2O5。该材料的层间距可达9.50 Å, 有利于K+的可逆嵌脱。X射线衍射结果显示:当材料完全放电至1.5 V时, 其层间距缩小为9.06 Å;当充电至3.8 V时, 材料的层间距恢复为9.50 Å。值得注意的是, SEM图谱显示充放电过程中重复的晶格膨胀与收缩并没有造成材料结构的坍塌。在100 mA/g的电流密度下, 循环250周后仍能保持81%的初始容量。通过X射线光电子能谱的结果可以分析出, 在钾离子的嵌脱过程中, 钒离子的价态是在+4和+5价之间可逆转换。为了研究K0.5V2O5材料在嵌脱钾时的热力学和动力学机理, 研究者进行了恒电流电歇滴定测试, 结果表明K+在K0.5V2O5中的扩散对材料嵌脱钾过程中的动力学起到了决定性作用, 这意味着可以通过调整活性材料的微观结构进一步增强其电化学性能。
另一种氧化物K0.69CrO2材料在研究中也表现出较为优异的电化学性能, Sun等[24]采用电化学离子交换法从O3-NaCrO2中成功地合成了纯相P3-K0.69CrO2。材料在0.1 C倍率下显示出100 mAh/g的高可逆容量, 即使在1 C倍率下循环1000圈仍能保持65%的初始容量。
2.3 聚阴离子类正极材料
聚阴离子正极材料具有开放性的三维框架结构、强诱导效应和X-O强共价键, 因此其作为钾离子电池正极材料具有离子传输快、工作电压高、结构稳定等优点。聚阴离子型化合物的通式为KxM(XO4)3(M是V、Ti、Tr、Al、Nb等或其中的几种组合;X是P或S;0≤x≤4)。M多面体与X多面体通过共边或者共点连接而形成多面体框架, 而K+位于框架间隙中。由于X 四面体的诱导效应, 该类材料中的过渡金属Mn+具有较高的氧化还原电对。
LiFePO4已广泛应用于锂离子电池正极材料, 虽然有报道称少量的K可以占据Li位点, 但是由于钾离子尺寸较大, 制备纯橄榄石相KFePO4仍然是一个巨大的挑战。Mathew等[25]提出将无定形的FePO4作为钾离子电池正极材料, 无定形FePO4具有的短程有序结构可以促进客体离子K+的插入。电化学测试表明在1.5~3.5 V电压范围内, 每个FePO4单元可以容纳0.89 K+, 这相当于156 mAh/g的高放电容量。非原位XRD结果显示非晶相FePO4材料在充放电过程中发生了从无定形到结晶相的可逆转变。这项研究工作对于开发类似的过渡金属磷酸盐电极材料提供了可能性。
近年来钒基聚阴离子型材料作为储钾电极材料受到广泛关注, Xu等[26]首次提出将快离子导体NASICON型的K3V2(PO4)3/C复合物作为钾离子电池阴极材料。在2.5~4.3 V电压窗口内该材料可以提供54 mAh/g的可逆容量, 其放电平台在3.6~3.9 V内, 在20 mA/g的电流密度下循环100周后能保持初始容量的80%。他们通过将纳米K3V2(PO4)3/C复合材料与块状的K3V2(PO4)3相比较, 发现K3V2(PO4)3/C复合物具有更有利的储钾主体结构。其3D多孔的结构有利于K+的扩散, 原位碳涂层保证了反复的充放电过程中结构的稳定性且促进了固体电解质界面膜SEI的形成。此外, 纳米级K3V2(PO4)3颗粒更容易与电解液充分接触, 从而使得活性物质积极参与到电化学反应中。
研究表明通过减小材料尺寸、表面碳包覆和元素掺杂等方式可以有效提升钾离子电池聚阴离子型正极材料的电化学性能。Komaba等[27]在聚阴离子型框架结构中引入强电负性的F和O, 制备出具有4 V工作电压平台的KVPO4F 和 KVOPO4正极材料。在2.0~5.0 V的电压窗口内, 前者由于结构中的V(Ⅲ)O4F2八面体而存在V3+/V4+氧化还原电对, 在4.13 V的工作电压平台下可以提供92 mAh/g放电容量;后者的V(Ⅳ)O6八面体表现出中心离子V4+/V5+的氧化还原反应, 在4.0 V的工作电压平台下放电容量可达84 mAh/g。通过在结构中引入强吸电子基团, 提高了材料的工作电压平台, 从而使材料的能量密度得以提升。Ceder等[28]采用两步固相法合成了化学计量的KVPO4F正极材料, 电化学测试结果表明其具有极高的放电电压4.33 V, 可提供105 mAh/g的比容量和454 Wh/kg的质量能量密度。为了更好地理解KVPOF中氟氧比例对其电化学性能的影响, 研究者通过温和的还原环境制备出KVPO4.36F0.64。他们指出KVPO4F的氧化导致阴离子的无序结构, 造成更加平缓的电压平台。同时, F含量的降低使诱导效应减弱, 电压平台下降。另一方面, 氧含量的增加使得V的初始价态升高, V的氧化还原反应受到限制, 从而降低了材料的比容量。O少量取代F虽然造成容量的衰减, 但是材料的循环性能和倍率性能却得到提高。基于Na3V2(PO4)2F3的研究, Zhang等[29]报道了一种新型的氟磷酸盐K3V2(PO4)2F3阴极材料, 在3.7 V的工作电压下可以给出100 mAh/g的比容量。将材料在10~100 mA/g的电流密度下进行电化学测试, 发现仍有80%的初始容量, 即使在2 C倍率下也有50 mAh/g的容量。优异的倍率性能归因于VO4F2八面体和PO4四面体构建的开放框架结构。值得一提的是, 这里制备的K3V2(PO4)2F3具有约为2 μm的较大尺寸, 通过控制颗粒尺寸和碳涂层的方式可以进一步提高材料的倍率性能。同时, 这项工作证实了从锂电和钠电类似物中寻求有潜力的电极具有重大意义, 同时也促进了高性能钾离子电池正极材料的发展。
2.4 有机化合物类正极材料
有机化合物类正极材料结构设计灵活、理论容量高、环境友好、价格低廉, 是一类具有广阔应用前景的储钾材料。Hu等[30]提出将对菲四甲酸二酐(PTCDA)作为非水性钾离子电池正极材料, PTCDA电极材料在1.5~3.5 V的电压窗口内可以表现出131 mAh/g的比容量, 在50 mA/g的电流密度下循环200周后容量保持率为66.1%。当设置放电截止电压为0.01 V时形成了K11PTCDA, 可以提供753 mAh/g的容量, 表明该材料也可作为高容量的钾离子电池负极材料。Ji等[31]证明了KPTCDA作为钾离子电池正极材料时, 在20 mA/g的电流密度下能够给出122 mAh/g的容量。为了更好地理解该材料的储钾机制, 研究者采用了非原位XRD和非原位红外光谱技术。分析结果显示:材料在嵌钾的过程中经历了高度的非晶化, 脱钾的过程中有序结构在2.8 V时会发生部分的扭曲, 这种现象表明非晶化是部分可逆的。
醌类化合物电极材料由于具有接近600 mAh/g的理论容量, 高的氧化还原电位和良好的可逆性等优势, 已在储锂和储钠电极材料中得到广泛研究。Ji等[32]通过菲利普斯方法(Phillips method:简单的缩聚反应)对1, 5-二氯蒽醌进行聚合得到了1, 4-苯醌聚合物(PAQS)。该聚合物作为钾离子电池正极材料可以提供190 mAh/g的可逆容量, 研究者注意到电池隔膜在循环50周后变成了深黄色, 表明部分PAQS溶解在电解液中, 造成容量略有衰减。
对于正极材料来说, 普鲁士类材料具有优异的电化学性能, 展现出广阔的应用前景;层状过渡金属氧化物虽给出了最高的理论比容量, 但是长期循环过程中容量衰减严重, 通过电化学惰性阳离子的取代, 可以增强此类材料的结构稳定性;聚阴离子型材料兼具电压平台高和循环稳定性好的特性, 但是该类材料需要解决导电性差的问题。
3 钾离子电池负极材料
3.1 嵌入类负极材料
嵌入型钾离子电池负极材料主要包括两种:碳基负极材料和钛基负极材料。其中最典型的嵌入类负极材料就是碳材料, 碳负极材料在安全和循环寿命方面都表现出良好的性能, 而且成本低廉、环境友好。根据材料石墨化程度的不同, 碳基负极材料通常可以分为石墨、软碳和硬碳等。石墨晶体具有平面六边形的网状结构, 这些网状结构以范德华力构成互相平行的层状结构, 层间距为0.354 nm。由于其同层的sp2杂化碳原子中形成离域化的π键电子在石墨层间能够自由移动, 使得石墨具有良好的导电能力。石墨类材料独特的结构非常适合锂离子的反复嵌脱, 是目前应用最为广泛、技术最为成熟的锂电负极材料, 其理论容量可达372 mAh/g。
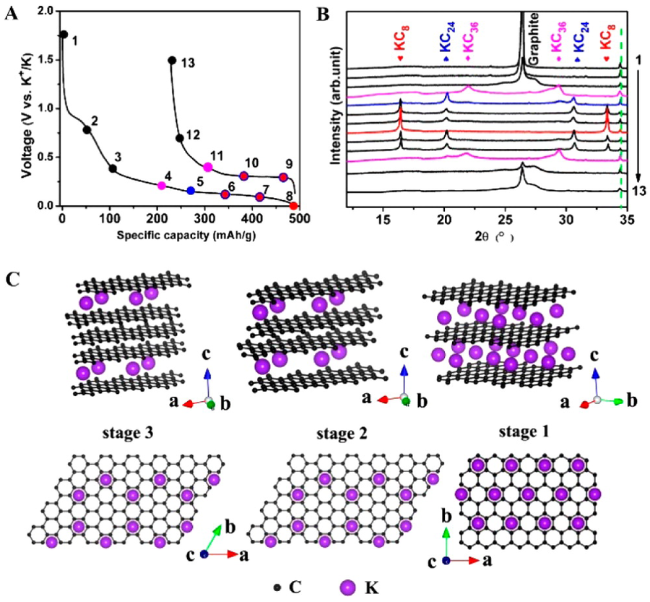
Ji等[33]和Komaba等[34]证实了钾离子可以通过电化学方法嵌入到石墨中, 可逆容量可达273 mAh/g。通过非原位XRD分析结果表明钾离子在嵌入石墨负极的过程中存在着这样的阶变:C→KC24→KC16→KC8。Hu等[35]将还原氧化石墨烯(rGO)作为钾离子电池负极材料, 具有208 mAh/g的可逆比容量。Wang等[36]研究了石墨负极材料在钾离子电池中的电化学性能, 他提出电解液和黏结剂的选择对材料的电化学性能至关重要。当海藻酸钠作为黏结剂, 碳酸乙烯酯(EC)与碳酸丙烯酯(PC)作为电解液时, 石墨负极展现出更为优异的电化学性能, 可提供246 mAh/g的可逆比容量, 且循环200周后容量还有89%。
石墨作为钾离子电池负极材料时具有较高的可逆容量与良好的倍率性能, 但由于钾离子尺寸较大, 影响其在材料中的扩散速率;同时, 电极材料在循环期间体积变化大, 会造成容量的快速衰减。为了解决石墨负极材料出现的扩散速率差和体积膨胀等问题, 研究人员采取了一系列的改性工作。Share等[37]将氮掺杂石墨烯作为钾离子电池体系的负极材料, 在50 mA/g的电流密度下, 氮掺杂石墨烯的钾储存容量可达350 mAh/g。他们通过原位拉曼光谱技术观察G带的偏移现象, 探究了不同的脱钾阶段氮原子对储钾性能的影响。分析结果表明, 氮掺杂维持了KC8的形成并提供了额外的电荷容量, 而且还能激活石墨晶格中的储钾位点, 从而提高了材料的储钾能力。Ju等[38]通过热退火方法合成了磷氧双原子共掺杂的石墨烯材料, 并将其应用为钾离子电池负极材料。在50 mA/g的电流密度下循环50周后比容量高达474 mAh/g, 在500 mA/g的大电流密度下充放电600周后可给出385 mAh/g的比容量。他们指出优异的电化学性能归因于杂原子掺杂后形成的特殊结构。磷和氧双重掺杂增大了材料的层间距, 有利于钾离子在石墨烯层间的传输。掺杂磷氧原子后形成的连续薄膜网状结构能够促进电子的快速传输, 提高了石墨烯材料的电导率。同时, 磷氧原子掺杂形成的空位和缺陷可有效地缓冲石墨烯材料在循环过程中的体积变化, 且能够为钾离子的嵌脱提供丰富的活性位点。Mai等[39]将冷冻干燥得到的海绵状氧化石墨烯进行硫化和还原制备出硫掺杂的还原氧化石墨烯(S-RGO)海绵。S-RGO负极材料在50 mA/g的电流密度下循环150周后可提供361 mAh/g的储钾容量。他们通过拉曼光谱分析表明硫掺杂形成的畸变结构可以有效提高石墨烯的储钾性能。掺杂硫后, S-RGO海绵的含氧官能团减少, 从而降低了SEI形成过程中不可逆的K+消耗, 提升了库仑效率。与常规的C位点相比, 噻吩类S位点可以提供更多的空间来减少充放电过程中的体积膨胀并降低K+吸附能。总之, 掺杂硫的氧化还原石墨烯可以有效提高K+吸附能力, 提高比容量, 增强长循环稳定性。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
图4 (A)在C/10电流密度下的第一圈恒流充放电曲线;(B)第一圈循环中选定荷电状态的非原位XRD表征;(C)钾离子嵌入石墨后形成的钾层间化合物的结构示意图[33]Fig. 4 (A) First-cycle galvanostatic charge and discharge profiles at C/10;(B) Ex situ XRD for selected states of charge in the first-cycle;(C) Structure diagrams of different K-GICs, side view(top row) and top view(bottom row)[33].Copyright 2015 American Chemical Society |
软碳(SC)是一种经低温碳化后得到的负极材料, 它们主要由石墨微晶构成。软碳结晶度低, 与电解液的相容能力强, 且层间距较大, 有利于钾离子的快速嵌脱。Ji等[33]通过热分解有机芳香族化合物3, 4, 9, 10-苝四酸二酐(PTCDA)制备出非石墨化的软碳材料, 其密度为1.6 g/cm3, 低于石墨的密度2.3 g/cm3。与石墨负极快速的容量衰减不同, 软碳则表现出大大提高的循环稳定性, 在2 C的倍率下循环50周后容量保持率为81.4%。
硬碳(HC)是指难以石墨化的碳, 是高分子聚合物的热解碳。相比传统的石墨负极材料, 硬碳作为钾离子负极材料具有很多优势。硬碳具有各向同性的结构, 层间距较大, 有利于钾离子在结构中的自由扩散;疏松多孔和相互交错的类石墨插层状结构, 有丰富的储钾位点;硬碳材料具有更高的可逆比容量, 良好的低温性能和优异的倍率性能。Ji等[40]以蔗糖作为原料, 采用水热法合成了硬碳微球。他们指出其作为钾离子电池负极材料比作为钠离子电池负极具有更好的电化学性能。当硬碳微球作为钾离子电池负极材料时, 可以提供262 mAh/g的比容量, 即使在2 C和5 C的大倍率下, 容量仍有190 mAh/g和136 mAh/g。钾离子的反复嵌脱不会破坏硬碳的结构, 在循环100周后容量保持率可达83%。
有序介孔碳材料(OMC)是指孔径分布在2~50 nm之间的多孔碳材料。Guo等[41]将非晶介孔碳作为钾离子电池负极材料进行电化学测试, 发现其比一般的晶体石墨碳具有更好的性能。在0.05 A/g的电流密度下, 非晶介孔碳的比容量可达257.4 mAh/g。即使在1.0 A/g的大电流密度下循环1000周后, 仍可提供146.5 mAh/g的容量。非晶介孔碳经过完全充放电之后, 形成了短程有序的结构, 层间距分别为4.18 Å和4.53 Å。在充放电过程中, 这种较大的层间距有利于钾离子的自由嵌脱, 同时缓解了钾离子嵌入后引起的层间距的不可逆增加和体积膨胀。非晶有序介孔材料的多孔性可以使大量的钾离子吸附在材料边缘和缺陷处的活性位点上, 独特的结构特征使非晶介孔碳材料表现出较高的比容量和出色的长期循环稳定性。Wan等[42]以酚醛树脂为原料合成了类神经元结构的中空碳骨架, 有趣的是, 材料在碳化的过程中其骨架会形成内部中空的玻璃泡结构。当用作钾离子电池负极材料时, 具有互连中空通道的碳骨架可以为反复的充放电过程提供一个弹性结构。在0.1 C倍率下能够提供340 mAh/g的高比容量, 即使在0.5 C倍率下也可稳定循环超过150次。
钛基负极材料作为钾离子电池负极材料时, 也是通过嵌入类反应机制进行储钾的。Kishore等[43]采用固相法合成了单斜晶系钛酸盐K2Ti4O9, 在30 mA/g的电流密度下其初始放电容量为97 mAh/g。Xu等[44]通过水热法制备出纳米立方相钛磷酸盐KTi2(PO4)3。当被用作钾离子电池负极材料时, KTi2(PO4)3表现出74.5 mAh/g的初始放电容量, 钛基材料的带隙宽, 电子导电能力差, 第二周循环开始放电容量衰减严重。随后他们采用碳包覆对材料改性, KTi2(PO4)3/C的首圈放电容量可达75.6 mAh/g, 即使在循环100周后仍有很好的容量保持率。Wu等[45]经碱化处理合成了Ti3C2 Mxene纳米带(a-Ti3C2 MNRs), 由于层间距的扩大和三维多孔结构的存在, a-Ti3C2 MNRs可实现快速的电子传输和离子扩散。该碳化钛化合物作为钾离子负极进行电化学测试, 其在20 mA/g的电流密度下可以给出136 mAh/g的可逆容量;在200 mA/g的高电流密度下循环500圈后仍可保留42 mAh/g的可逆容量。
3.2 合金类负极材料
合金类储钾负极材料是指能和钾发生合金化反应的金属及其合金、中间相化合物及复合物。由于发生的是多电子的合金化反应, 合金类负极材料具有较高的理论容量。然而高的合金化容量同时会造成循环过程中巨大的体积变化, 由此带来的结构应力会导致活性物质粉化与脱落, 丧失与集流体的电接触, 从而引起电极容量的快速衰减。目前钾离子电池合金类负极材料主要集中于锡(Sn)基、锑(Sb)基和磷(P)基合金型储钾材料。
金属锡直接被用作钾离子电池负极材料时, 在锡与钾的合金化过程中会产生严重的体积膨胀问题, 因此需要对其进行改性。Glushenkov等[46]采用机械球磨法将Sn纳米颗粒与石墨混合制备出Sn-C复合材料。利用碳材料作为缓冲体系, 不仅缓解了锡基负极材料的体积效应和纳米颗粒的团聚问题, 又有利于锡基材料表现出高容量的特点。在25 mA/g的电流密度下Sn-C复合物可以提供150 mAh/g的可逆比容量。Zhang等[47]将Sn4P3/C复合物作为新型的钾离子电池负极材料, 在电流密度50 mA/g下可提供384.8 mAh/g的可逆容量, 在1 A/g电流密度下也具有良好的倍率性能, 可以给出221.9 mAh/g的容量。且其放电平台为0.1 V, 避免了因钾枝晶的生长引起的内部短路的风险。他们指出充放电过程中K-Sn和K-P合金的形成为体积变化提供了缓冲区, 碳基质也可作为体积膨胀的缓冲剂, 同时起到稳定结构和增加导电性的作用。
锑的合金型材料的理论嵌钾容量高达660 mAh/g, 且锑与钾的合金化过程中具有平坦的电压平台, 能够提供稳定的工作电压;金属锑具有较高的电导率, 可促进电子的传输和转移。Wu等[48]证明了锑-碳复合材料在嵌脱钾的过程中能和钾发生可逆反应, 生成K3Sb合金, 其可逆容量高达650 mAh/g。
磷可与碱金属K形成多种化合物, 其理论储钾容量高达2595 mAh/g, 且在离子电池中具有安全的反应电极电位。但是磷本身的电子电导性差, 循环过程中体积会出现巨大的变化, 致使其循环稳定性恶化, 从而很大程度上限制了磷基负极材料在钾离子电池中的应用。最近, Xu等[49]通过蒸气-转化的方法将红磷纳米颗粒锚定在3D多孔的碳纳米片框架中, 利用碳材料的电子导电性和结构稳定性, 实现了红磷在钾离子电池中的长期循环性能和倍率性能的突破。在100 mA/g电流密度下可以达到655 mAh/g的高可逆容量, 并且具有良好的倍率性能, 在电流密度2000 mA/g下仍有323.7 mAh/g的容量。纳米化的红磷颗粒能够减小其表面的应力, 同时减小钾离子的扩散距离;3D多孔的碳纳米片框架保证纳米颗粒充分分散并且颗粒间留有足够空间, 以缓冲体积的膨胀。
3.3 转化类负极材料
与钾离子的嵌脱反应完全不同, 转化反应实质上就是置换反应。过渡金属硫族化合物MSx都是基于转化类反应储能机理的钾离子电池负极材料。由于转化类负极材料发生的是多电子反应, 因此具有较高的比容量和能量密度。
Jiao等[50]通过静电纺丝和随后的热处理法制备了嵌于多孔氮掺杂碳纳米纤维的V2O3(V2O3@PNCNFs)柔性自支撑薄膜电极材料, 将其作为钾离子电池负极材料时, 在50 mA/g电流密度下可表现出240 mAh/g的可逆比容量, 循环500周后容量保持率高达95.8%;当电流密度增大到1000 mA/g时, 仍具有134 mAh/g的充电比容量。研究者通过电化学动力学分析、原位XRD以及密度泛函理论计算等发现V2O3的主要储钾机理为嵌入赝电容效应。对于嵌入赝电容效应机理, V2O3在储钾过程中没有发生相的转变, 从而使得其在电化学反应中保持了超稳定的结构。这在钾离子电池中是首次报道, 同时此项研究工作为开发用于快速储能的赝电容高效纳米电极材料提供了研究思路。
Rahman等[51]将Co3O4-Fe2O3纳米颗粒分散在超导炭黑制备出复合材料作为钾离子电池负极材料, 其在50 mA/g电流密度下循环50周后可展现出220 mAh/g的可逆容量。过渡金属硒化物具有较高的储钾容量和合适的反应电位, 得到研究者们的广泛关注。Lu等[52]设计合成了层间距为0.678 nm的碳氮掺杂MoSe2, 基于1 M KFSI/EMC电解质, MoSe2/N-C负极材料表现出优异的电化学性能, 在0.1 A/g电流密度下, 循环300周后容量高达258.02 mAh/g。他们通过非原位XRD、Raman和HRTEM技术分析其储钾机理, 发现MoSe2/N-C先经历嵌钾反应, 然后进行转化反应最终生成K5Se3。最近, Yu等[53]制备出氮掺杂碳纳米管负载的CoSe2, 每个金属八面体CoSe2颗粒沿着碳纳米管依次排列, 并且锯齿形空隙可以缓冲循环期间的体积膨胀, 从而提高了长期循环稳定性, 将其应用于钾离子电池负极材料, 获得了优异的电化学性能。这种独特的结构在电流密度0.2 A/g下循环100周后, 可提供253 mAh/g的高容量。即使在2.0 A/g的电流密度下循环600圈, 容量仍有173 mAh/g, 相当于每圈只有0.03%的容量衰减。
对于负极材料, 嵌入类负极材料循环性能较好, 但是通常表现出较低的可逆容量, 通过微观结构调控和元素掺杂等方法来提高此类材料的可逆容量;合金类和转化类负极材料可提供较高的理论容量, 然而在充放电中体积膨胀严重, 采用纳米化、掺杂、包覆等手段来缓冲钾离子嵌脱过程中的体积变化也是该类材料研究的主要方向。
4 钾离子电池电解液
电解液作为连接钾离子电池正极和负极的盐桥, 是钾离子电池体系关键的组成部分, 直接影响着电池的电化学性能和安全性能。电解液不仅可以补充离子, 加速离子传导等, 而且其分解电压决定了钾离子电池的工作电压窗口、电导率和使用温度等。通过对电解液体系的优化和选择, 能够提高电池的能量密度。对于非水系钾离子电池, 其电解液主要是以无机钾盐为溶质, 有机碳酸酯类或醚类为溶剂的溶液。常用的电解质盐有:高氯酸钾(KClO4)、双三氟甲烷磺酰亚胺钾(KTFSI)、六氟磷酸钾(KPF6)和双氟磺酰亚氨钾(KFSA)等。电解液溶剂基本采用碳酸乙烯酯(EC)、碳酸丙烯酯(PC)、碳酸二甲酯(DMC)、碳酸二乙酯(DEC)和乙二醇二甲醚(DME)等, 实际应用中为了满足高离子电导率、宽电化学窗口、高机械强度以及电化学和热稳定性等要求, 一般采用二元组合, 例如EC+PC和EC+DEC等。由于有机电解液很容易腐蚀钾金属电极, 影响电池的电化学性能, 因此通常在其中加入成膜添加剂氟代碳酸乙烯酯(FEC)来改善。
Xu等[36]研究了石墨负极在三种不同电解液(EC∶PC、EC∶DEC以及EC∶DMC)中的电化学性能差异。结果显示当采用KPF6∶EC∶PC电解液时, 首圈库仑效率最高, 且在很快的时间内库仑效率会稳定在100%左右。而采用后两种电解液时, 首圈库仑效率很低, 可能是由于DEC和DMC在低电压下不稳定造成容量不可逆。尤其是采用KPF6:EC:DMC电解液时, 半电池循环的整个过程中其库仑效率始终低于90%且在循环70周后库仑效率快速下降, 表明SEI膜不断地形成, DMC不断地消耗分解。可见, 选择合适的电解液, 形成稳定的SEI膜, 对电池的电化学性能稳定性至关重要。
Komaba等[54]探究了不同的电解质盐和添加剂对电池电化学性能的影响。KFSI盐在EC∶DEC (1∶1 v/v)电解液溶剂中的溶解度为 1.0 mol/dm3, 高于KPF6盐在其中的溶解度。但是在高电压下, Al会溶解于KFSI盐中, 造成充电过程中出现不可逆的现象。将KFSI盐换作KPF6盐, 并且加入2 vol% FEC添加剂后, 以K2Mn[Fe(CN)6]为正极组成的半电池显示出出色的库仑效率和稳定的循环性能。他们指出FEC分解后形成稳定的SEI, 抑制电解质的进一步分解, 新型电解液添加剂对高性能的钾离子电池发展的影响不容小觑。He等[12]也证实了在以普鲁士白化合物K1.7Fe[Fe(CN)6]0.9为正极材料的钾离子电池中, 电解液中加入FEC添加剂后, 半电池的库仑效率和长循环稳定性显著提高。然而, Dugas等[55]指出FEC添加剂作用于负极材料时会产生很大的极化现象。
溶剂的选择对提高电解液的润湿性和电池的性能至关重要。Chen等[56]合成了对苯二甲酸钾(K2TP)有机负极材料, 并采用DME作为溶剂, 相应的DME基电解液可以在K2TP表面形成稳定的SEI膜, 且电压平台为0.6 V, 避免了钾枝晶的生成。为了探究DME基电解液中SEI膜对K2TP电极性能的关键作用, 他们对比了K2TP在DME基电解液与碳酸酯类电解液中的电化学性能, 充放电过程中在碳酸酯类电解液中形成的SEI膜比DME基电解液中的SEI膜更厚, 从而导致高的电荷转移电阻和低的库仑效率。对于充放电过程中电解液与活性物质间的相互作用, 有待进一步深入研究。
本文将一些代表性的研究工作及其相关材料的电化学性能进行了汇总(详细见表1)。
表1 近年来关于钾离子电极材料电化学性能汇总表Table 1 Summary of electrochemical performance about potassium-ion electrode materials in recent years |
| Molecular formula | Electrochemical property | ref | |||
|---|---|---|---|---|---|
| Voltage range (V vs K/K+) | Current | Initial capacity (mAh·g-1) | Cycle performance | ||
| K0.220Fe[Fe(CN)6]0.805·4.01H2O | 2.0~4.0 V | 50 mA·g-1 | 76.7 | 93.4%(50 cycles) | 2 |
| K1.92Fe[Fe(CN)6]0.94·0.5H2O | 2.0~4.3 V | 0.1 C | 133 | 92.8%(200 cycles) | 4 |
| K1.89Mn[Fe(CN)6]0.92·0.75H2O | 2.5~4.6 V | 0.2 C | 142.6 | — | 5 |
| K1.75Mn[Fe(CN)6]0.93·0.16H2O | 2.0~4.5 V | 30 mA·g-1 | 137 | — | 8 |
| KFeⅡ[FeⅢ(CN)6] | 2.0~4.5 V | 10 mA·g-1 | 118.7 | 93.73%(100 cycles) | 10 |
| K1.7Fe[Fe(CN)6]0.9 | 2.0~4.5 V | 10 mA·g-1 | 140 | 65%(300 cycles) | 11 |
| P2-K0.6CoO2 | 1.7~4.0 V | 10 mA·g-1 | 82 | 87%(300 cycles) | 16 |
| K0.7Fe0.5Mn0.5O2 | 1.5~4.0 V | 20 mA·g-1 | 178 | 76%(250 cycles) | 19 |
| K0.65Fe0.5Mn0.5O2 | 1.5~4.2 V | 20 mA·g-1 | 151 | 78%(350 cycles) | 20 |
| V2O5·0.6H2O Xerogel | 1.5~4.0 V | 50 mA·g-1 | 224.4 | 78.3%(100 cycles) | 21 |
| P3-K0.69CrO2 | 1.5~3.8 V | 0.1 C | 100 | 65%(1000 cycles) | 23 |
| K3V2(PO4)3/C | 2.5~4.03 V | 20 mA·g-1 | 77 | — | 25 |
| KVPO4F | 2.0~5.0 V | 0.05 C | 92 | 97%(30 cycles) | 26 |
| KVPO4F | 3.0~5.0 V | 5 mA·g-1 | 105 | — | 27 |
| K3V2(PO4)2F3 | 2.0~4.6 V | 10 mA·g-1 | 100 | 97%(100 cycles) | 28 |
| PTCDA | 1.5~3.5 V | 10 mA·g-1 | 131 | 66.1%(200 cycles) | 29 |
| Poly(anthraquinonyl sulfide) | 1.5~3.4 V | 20 mA·g-1 | 211 | 75%(50 cycles) | 31 |
| Graphite | 0~1.5 V | 20 mA·g-1 | 246 | 89%(200 cycles) | 35 |
| Nitrogen-Doped Graphene | 0~1.5 V | 50 mA·g-1 | 350 | — | 36 |
| S-RGO | 0.01~3.0 V | 50 mA·g-1 | 3648 | 79%(50 cycles) | 38 |
| Hard Carbon Microspheres | 0~1.5 V | 28 mA·g-1 | 262 | 83%(100 cycles) | 39 |
| Ordered mesoporous carbon | 0.01~2.6 V | 50 mA·g-1 | 307.4 | — | 40 |
| Red P@CN | 0.01~2.0 V | 100 mA·g-1 | 715.2 | — | 48 |
| MoSe2/N-C | 0.01~3.0 V | 100 mA·g-1 | 278.3 | — | 50 |
5 结论
近年来, 为应对环境危机和能源危机, 新能源领域的发展如火如荼。其中, 钾离子电池得益于钾元素储量丰富、成本低廉, 且物理化学性质与锂离子相似, 在离子电池领域中具有辉煌的应用前景。目前, 正极材料主要集中于普鲁士蓝类材料, 但具有较高理论比容量的层状过渡金属氧化物正极与兼具高电压和稳定性的聚阴离子正极材料也有一定程度的研究;负极材料则主要集中于嵌入型和合金型负极材料, 虽然各自具有一定程度的缺陷, 但是经过掺杂、包覆、纳米化等手段改性后, 具有较为优异的电化学性能;电解液方面则是在与材料的匹配性上进行了部分探索。尽管受限于K+尺寸因素, 相比于锂离子电池, 钾离子电池具有一定不足, 但随着科研者不断深入和广泛的研究, 钾离子电池已经展现出其光辉的未来。