




1 引言
化学氧化技术可以破坏有机污染物的分子结构,并可将其彻底矿化[7],其中,先进(高级)氧化法(advanced oxidation processes,AOPs)或称先进(高级)氧化技术(advanced oxidation technologies,AOTs)[8]因其对难降解有机污染物的去除效果好而受到广泛关注[9]。目前常见的AOPs中,基于羟基自由基(·OH)的Fenton类氧化法,因其所产生的具有强氧化能力的·OH可降解并矿化难降解有机物而较早受到关注[10],但该方法有试剂用量大,对氧化对象无选择性,氧化效率低且反应需要强酸条件(pH 3~4)[11]等缺点;臭氧类氧化法耗能高、水溶解度低,且臭氧不稳定易在空气中分解[12];超临界水氧化法操作条件苛刻,设备要求高,同时存在腐蚀和盐沉淀问题[13];而基于硫酸根自由基(SO4.-)的AOPs虽然早在1986年就有报道,但其用于污染物的降解研究却始于1996年[14]。近年来,作为一种新兴的AOPs,其氧化机理、活化方法及其在新型有机污染物降解上的应用逐渐成为一个研究热点。
SO4.-主要由过一硫酸盐(peroxymonosulfate,PMS)或过二硫酸盐(peroxydisulfate,PDS)等过硫酸盐(persulfate,PS)活化产生。有的学者将过二硫酸盐也称为persulfate(PS)[21,22]。PMS是不对称结构的白色固体粉末,溶解度为250 g/L,水溶液呈酸性,pH值在小于6或等于12时较稳定,但当pH值为9左右时,其稳定性很差,几乎一半的HSO5-分解为SO52-[17,23],常用的PMS为过一硫酸氢钾复合盐(2KHSO5·KHSO4·K2SO4);PDS是对称结构的无色或白色结晶,具有较高的稳定性,易溶于水,溶解度为730 g/L,水溶液也呈酸性,常用的PDS为Na2S2O8和K2S2O8[15]。PDS成本低于PMS[19],水溶性和室温下稳定性更高[24],且由于PDS的O-O键的键能(33.5 kcal/mol)低于PMS,所以PDS对污染物的降解率更高。Shah等[25]对比了UV/PMS、UV/PDS和UV/H2O2三种体系降解硫丹(endosulfan)的效果,相同条件下UV/PDS降解率最高(91%),其次是UV/PMS(86%),而UV/H2O2则最低(64%),这是因为PDS的O-O键的键能(33.5 kcal/mol)远低于H2O2(51 kcal/mol),而PMS则介于以上两者之间。
PS自身在室温下较稳定,对有机物的氧化速率较低,氧化效果不明显,但经活化后产生的SO4.-氧化效果有极大的提升。Chen等[26]对头孢氨苄的降解研究显示,只加PDS时,几乎不发生降解,而加入Cu活化PDS产生SO4.-后,头孢氨苄被完全降解。
2 PS的活化方法
PS的活化方法包括热活化(thermal activation)、机械化学活化(mechanochemical activation)、过渡金属活化(transition metal activation)、碳质材料活化(carbonaceous materials activation)、碱活化(alkali activation)、紫外活化(ultraviolet(UV)activation)和电化学活化(electrochemical activation)等。不同方法的活化效果各异,对不同性质和状态污染物的适用性也不同,因此在实际应用中,不同的污染物应根据其特点选择适当的活化方法。
2.1 热活化法
热活化是最简单的活化方法,无需添加外来物,避免了二次污染。热活化主要用于PDS的活化(易于PMS)[27],热活化的机制为:
S2O82-+heat→2SO4.-
HSO5-+heat→SO4.-+·OH
适度提高热活化温度可加快PS分解产生SO4.-的速率,从而提高有机污染物的降解率,但温度并非越高越好。Wang等[28]热活化PDS降解多环芳烃(polycyclic aromatic hydrocarbons,PAHs),60 ℃以下时,降解率随温度上升而提高;但超过60 ℃时,由于氧化剂的快速消耗,降解率反而降低。
由于热活化耗能,Ike等[29]用工业冷却水等废热活化PDS降解水中的腐殖酸,降解率达80%以上,但易受pH的影响。pH=8时降解率98%,而酸性条件时(2.5~7)则降至~50%,这主要受水中Cl-的影响,Cl-在酸性条件下与有机污染物竞争SO4.-,使降解率下降。总体而言,该方法不仅可降低能耗,还减少了废热排放造成的热污染,但缺点是反应速率较慢(120 h)且会受到水中Cl-、NO3-和CO32-等杂质离子的影响。
Hu等[30]用微波加热活化PDS降解对硝基苯酚,相比传统加热方式,可更有效地活化PDS并降低反应活化能。90 ℃时,微波加热的降解率(93%)远高于传统加热方式(35%)。此外,降解率不受pH(3~11)及杂质离子(Cl-、NO3-等)的影响,显示微波加热是一种非常有应用前景的热活化方法。
2.2 机械化学活化法
机械化学活化是将材料放入行星球磨机,通过磨球间的高速碰撞实现电子间的转换,并利用碰撞过程中产生的高温条件,使PS产生大量的SO4.-,降解固体污染物。机械化学活化的优点在于未添加其他物质,降低了添加剂副产物可能带来的环境风险,是一种绿色高效的活化方法。不同于其他方法,机械化学活化主要用于高浓度固体污染物的降解,但该活化方法需选择合适的研磨剂,操作复杂、成本较高,难以工程应用。
由于PDS的O-O键能低于PMS,所以PDS对污染物的降解率较PMS更高。Huang等[31]将PDS颗粒与粉状十溴二苯醚(BDE-209)加入球磨罐,3 h BDE-209降解率为99.6%,且总有机碳(total organic carbon,TOC)去除率为99.0%,低溴中间产物被SO4.-快速分解,未检测到中间产物。将研磨剂PDS换成PMS时,BDE-209的降解率降至67.8%,而使用氧化钙作为脱卤剂降解BDE-209,降解率仅51.7%。该方法不受pH的影响,但研磨剂与污染物的质量比(MPDS/EDF-209=40)、不同尺寸磨球的数量比(N10mm/20mm=4∶1)及球料比(M球重/BDE-209=50)对降解率有影响,过低或过高都会因表面接触未达最佳效果而影响降解率。
为排除碰撞过程中热活化的影响,Wang等[32]以PDS和Al2O3为研磨剂,机械化学活化PDS降解全氟辛基羧酸(perfluorooctanoic acid,PFOA),降解率达100%。其中Al2O3一方面通过共价键配位与PFOA结合,另一方面其表面羟基与SO4.-反应生成的·OH也参与降解。对照实验显示,仅加热PDS和Al2O3到100 ℃,2 h后PFOA无明显降解,证明只有发生碰撞才会引发分子的旋转、排列和重新的键合。
2.3 过渡金属活化法
过渡金属用于PS的活化时,有均相和非均相两种方式。均相金属活化时,Co和Ru是PMS最有效的活化剂,并且Co2+ > Ru3+ > Fe2+ > Ce3+ > V3+ > Mn2+ > Fe3+ > Ni2+[20],而Ag是PDS的最好的活化剂[33],但Ag成本高且对人体健康有害。V近年来才用于PS的活化,其机理有待进一步研究[34];而Cu在大多数情况下起协同作用,促进其他金属从高价态向低价态转变[35];Mn因环境友好而被广泛使用,但其催化活性较低,所以多与其他金属联用[36];Fe的研究最广泛,因为它对PMS和PDS均有不错的活化效果,成本低且浸出不像Co等其他金属一样危害环境和人体健康[20]。非均相金属活化既能消除单一金属的负面影响,又能通过离子间的协同作用提高降解率[36]。此外,一些框架(碳质材料或TiO2等)的构建也可避免金属离子浸出造成的环境危害[37]。金属活化PMS或PDS机制见图1 。
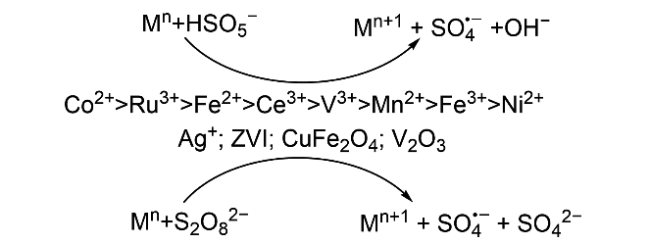
纳米级金属比表面积更大,与PS接触更充分,降解率更高。Deng等[38]用纳米零价铜活化PDS降解诺氟沙星(norfloxacin),5 min降解率达96%。Wang等[39]用NaBH4还原FeSO4·7H2O制备纳米零价铁(nZVI)活化PDS降解BDE-47,48 h降解率为64%,加入2.5 mM的Cu2+后降解率增至83%,因为Cu2+的高还原性将Fe3+还原为Fe2+去分解更多PDS,生成更多的SO4.-去降解目标污染物,但该方法后续要添加NaOH去除溶液中的Cu2+,且因nZVI颗粒间的强磁力和范德华力而易团聚,使其与PDS接触的点位减少,从而使SO4.-的产生量减少,最终导致降解率下降。Deng等[40]将Ca(OH)2包覆在nZVI颗粒表面,有效避免了以上问题,且Ca(OH)2不影响nZVI和PDS的接触。改性后的催化剂活化PDS降解磺胺二甲嘧啶(sulfamethazine),20 min降解率达100%,且不受地下水中低浓度Cl-、HCO3-和NO3-等离子的干扰。
表1 过渡金属活化PS降解有机污染物Table 1 Degradation of organic pollutants by transition metal activated PS |
| Class | Pollutant | Conc. (μM) | PS | Conc. (mM) | Activator | Conc. | pH | T(℃) | t(h) | kobs () | Degradation rate | ref | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| POPs | BDE-47 | 3.1 | PDS | 71.4 | 23,032∶1 | nZVI | 1000 mg/L | 6~11 | 25 | 48 | — | 64% | 39 |
| BDE-209 | 10.4 | PDS | 200 | 19,183∶1 | Fe2+ | 100 mM | 3~7 | — | 6 | 0.48 | 66% | 41 | |
| CB-28 | 3.9 | PDS | 2 | 500∶1 | V4+ | 1 mM | 5.5 | 25 | 25 | 0.03 | 80% | 34 | |
| phenanthrenea | 5.6 | PDS | 2 | 357∶1 | V2O3 | 100 mg/L | 3~5 | 25 | 3.6 | 7.15 | 88% | 42 | |
| PPCPs | carbamazepine | 21.2 | PMS | 1.66 | 78∶1 | Co3MnFeO6 | 200 mg/L | 3~10 | 35 | 0.5 | 10.98 | 100% | 36 |
| tetracycline | 225 | PDS | 5 | 22∶1 | Ag/AgCl/(0.05)FeX | 1000 mg/L | 3.5 | 25 | 2 | — | 100% | 43 | |
| norfloxacin | 15 | PMS | 1 | 67∶1 | Co0.4Fe2.6O4 | 200 mg/L | 6~8.5 | 40 | 0.05 | 122.40 | ~100% | 44 | |
| norfloxacin | 31.3 | PMS | 2 | 64∶1 | Co(Ⅱ)/TiO2 | 500 mg/L | 3~9 | 25 | 1 | 29.88 | ~100% | 37 | |
| ofloxacin | 27.7 | PMS | 2 | 72∶1 | Co(Ⅱ)/TiO2 | 500 mg/L | 3~9 | 25 | 0.5 | 73.20 | 100% | 37 | |
| Organic dyes | rhodamine B | 104.4 | PMS | 2 | 19∶1 | Co(Ⅱ)/TiO2 | 500 mg/L | 3~9 | 25 | 0.6 | — | ~100% | 37 |
| acid orange 7 | 56.9 | PMS | 0.55 | 10∶1 | Mn-Fe/LDH | 200 mg/L | 5~9 | 25 | 0.5 | 8.28 | 97.6% | 45 | |
| p-chloronitrobenzene | 2697 | PDS | 200 | 74∶1 | ZVI | 1 mmol/g | 3~6.8 | 25 | 8 | — | 90% | 46 | |
| Other pollutants | bisphenol A | 100 | PMS | 2 | 20∶1 | Mn/Fe3O4 | 200 mg/L | 7~11 | 15 | 0.5 | 43.20 | 100% | 47 |
| p-nitrophenol | 359.4 | PDS | 9 | 25∶1 | CuFe2O4 | 30 000 mg/L | 6 | 25 | 1 | — | 89% | 48 | |
| benzotriazole | 167.9 | PDS | 5 | 30∶1 | Cu(Ⅱ)/V2O5 | 200 mg/L | 9 | — | 2 | — | 97% | 35 |
a Phenanthrene is a type of polycyclic aromatic hydrocarbons(PAHs), which heretofore haven't been added to the POPs list in the Stockholm Convention. |
除POPs外,表1 中其他三类污染物都可以在室温下短时间内达到较高的降解率,而POPs的降解不仅时间长,降解率也较低,具体原因分析见3.1部分。由于过渡金属的存在,pH最佳范围随活化剂的特性而变化。如nZVI活化PDS降解BDE-47的最佳pH为6.5~11.5[39],这是由于强酸环境促使nZVI腐蚀并释放过量的Fe2+,过量的Fe2+与BDE-47竞争SO4.-,并会引起PDS分子中带负电的氧原子与H+结合,从而抑制其与带正电荷的Fe离子作用,使SO4.-产生量降低;而在碱性条件下生成的羟基可作为电子转移的活性中心,促进PDS的分解,从而提高降解率[39]。因此迫切地需要找到活化效果好、价格低廉并能适应不同环境的过渡金属。
2.4 碳质材料活化法
活性炭(activated carbon,AC)[49]、生物炭(biochar,BC)[50]等传统碳材料,以及有序介孔碳[51]、石墨烯[52]等新型纳米碳材料[53]一方面可作为金属的载体,另一方面因其较多的孔隙结构和丰富的官能团可吸附并活化PS产生SO4.-[54]。制备原料和条件不同,碳质材料的理化性质也不同,对PS的活化能力也各异。Liang等[55]以木屑为原料在800 ℃制备出AC,并用花生壳分别在400 ℃和700 ℃下制备出BC400和BC700,其中AC比表面积和孔隙体积最大,BC700次之,BC400最小。相同条件下三种碳质材料分别活化PDS降解磺胺甲口恶唑(sulfamethoxazde,SMX),虽然AC降解率(91.2%)大于BC700(88.7%)和BC400(30.4%),但在矿化程度上BC700要高于AC,这是由于AC主要把污染物吸附在表面降解(占总降解84%),而BC700在液相中的降解占比较大(53%)。BC可通过废弃秸秆厌氧燃烧产生,既节省了成本,一定程度上也解决了秸秆的回收利用问题,降解率也较高。如Wang等[56]通过玉米秸秆厌氧燃烧制备BC活化PDS降解诺氟沙星,50 min降解率为99.57%。但BC稳定性较差,二次重复使用降解率仅70.22%,第三次则低至40.84%,考虑到其来源绿色且价格低廉等优点,仍值得进一步研究。
碳质材料经表面化学改性可提高其吸附及活化PS的性能[53]。Pi等[57]用HNO3/H2SO4处理BC得到氧化BC(OBC),将BC和OBC分别与Fe3O4结合活化PDS,OBC对四环素(tetracycline)的吸附(50%)和降解(92.3%)均高于BC(30%和65.3%)。BC也可和nZVI等过渡金属协同促进PS的活化。但BC活化PS受到pH的影响,酸性和中性条件下降解率最佳,碱性条件下因生成·OH,降解率较低。由于BC灰分中富含Na+、K+、Mg2+和Ca2+等盐基离子,在土壤修复中会降低土壤氢离子及交换性铝离子水平,从而显著增加土壤的pH值[58],继而使降解率下降,因此BC活化法的工程应用有待深入研究。
2.5 碱活化法
碱活化PS的反应遵循下列途径[63]:
2S2O82-+2H2O→3SO42-+SO4.-+O2.- +4H+
SO4.- +OH-→SO42-+·OH
HSO5-+H2O→H2O2+HSO4-
H2O2+OH-→H2O+HO2-
HSO5-+HO2-→SO4.-+H2O+1O2
大量研究表明,pH对SO4.-氧化降解污染物的影响不容忽视。在酸性条件下,PS主要分解为SV,而中性和碱性条件下,SO4.-和·OH共存[64]。用碱活化PS时,需投加大量的碱以调整pH,这会增加成本并腐蚀设备。
2.6 UV活化法
在紫外光照射下,PS中的O—O键断裂,生成SO4.-。UV活化PS产生的自由基有两种来源,一种是UV照射直接产生,另一种是PS与被UV辐射的水分解生成的氢自由基反应间接产生。表2 总结了近年来UV活化PS降解有机污染物的一些报道,包括止痛药(anodyne)、降压药(antihypertensive drugs)、抗生素(antibiotics)及硫丹,同过渡金属活化一样,UV活化也受到pH的制约。
表2 UV活化PS降解有机污染物Table 2 Degradation of organic pollutants by UV activated PS |
| Class | Pollutant | Conc. (μM) | PS | Conc. (μM) | Wavelength(nm) | pH | t(h) | Degradation rate | ref | |
|---|---|---|---|---|---|---|---|---|---|---|
| Anodyne | antipyrine | 26.5 | PDS | 520 | 19.6∶1 | 253.7 | 7.2 | 1.00 | 80% | 69 |
| Antihypertensive drugs | atenolol | 20 | PMS | 80 | 4∶1 | 254 | 7 | 0.50 | ~80% | 70 |
| Antibiotics(sulfonamides) | sulfadiazine | 20 | PDS | — | — | — | 6.5 | 0.30 | 97% | 71 |
| Antibiotics (fluoroquinolones) | ciprofloxacin norfloxacin levofloxacin | 40 | PDS | 500 | 12.5∶1 | 254 | 6.5 | 0.16 | 42% 25% 1% | 67 |
| Antibiotics(β-lactams) | cephalexin oxacillin | 40 | PDS | 500 | 12.5∶1 | 254 | 6.5 | 0.16 | 84% 88% | 67 |
| POPs | endosulfan | 2.45 | PDS | 49 | 20∶1 | 254 | 7 | 1.30 | 90% | 25 |
Serna-Galvis等[67]用UV/PDS降解5种抗生素,相同条件下降解率差异较大(表2 ),各抗生素不同的化学结构决定了其光降解性各异。3种氟喹诺酮类抗生素环丙沙星(ciprofloxacin)、诺氟沙星和左氧氟沙星(levofloxacin)因其苯环不太亲电,不易受光取代的影响,因而光降解系数较低,导致其降解率较低;另一方面,ciprofloxacin、norfloxacin和levofloxacin向分子氧的能量转移量子产率分别为0.092、0.081和0.076[68],这表明ciprofloxacin诱导激发态氧分子的形成易于norfloxacin和levofloxacin。因此,ciprofloxacin可通过激发态氧的作用更快地降解,其降解率相对较高。
2.7 电化学活化法
电化学活化法是一种新型的PS活化方法,因其经济环保、过程可控及反应条件温和等优点而广受关注[72]。电化学活化PS降解污染物的机理比较复杂,主要依赖于阳极上直接和间接的氧化反应[73]。不同电极的氧化机理也有不同,Song等[74]认为掺硼金刚石(BDD)阳极上污染物的降解机理有直接电解、电解水以及电活化PS三种,其中电活化PS一方面可以促进电解水中·OH的生成,另一方面将PS转化为过渡态发生非自由基降解。自由基猝灭实验证明·OH在降解反应中起主要作用,其对抗癫痫药卡马西平(carbamazepine,CBZ)降解的贡献率分别为82.2%(PDS)和90.7%(PMS),这也是电活化PMS降解率高于PDS的原因。而Song等[72]用Ti/Pt电极完全降解CBZ需120 min,高于BDD为阳极的降解(20 min),因为Ti/Pt电极不产生·OH,所以在阳极上主要通过非自由基途径发生降解,而在阴极上PDS被活化产生SO4.-,但它对降解率的贡献小(30 min降解率38%)[75],所以目前电化学活化的研究主要集中在阳极反应上。
电化学活化也可与过渡金属联合,Cai等[24]用电化学活化PDS降解酸性橙(acid orange)时,向电解液中加入了以SBA-15(以SiO2为主体的有序介孔分子筛)为载体的Fe-Co,5 min降解率可达60%,而不加Fe-Co时仅30%,因为除电化学反应外,Fe-Co活化了PDS,且高价态Fe、Co离子在阴极被还原为低价态,再度作为催化剂活化PDS,从而产生更多的SO4.-参与反应。
综上,各活化方法适用范围不同,应根据实际情况选择适当的活化方法。表3 总结了不同活化方法的优缺点。
表3 各种PS活化方法的优缺点Table 3 Advantages and disadvantages of different PS activation methods |
| Activation method | Advantages | Disadvantages |
|---|---|---|
| Thermal | easy operation; no chemicals addition | high cost; poor stability |
| Mechanochemical | rapid reaction; no chemicals addition | suitable for solid pollutants |
| Transition metal | high degradation efficiency; easy operation; no special device and low energy consumption | metal ions leaching; poor stability and efficiency; high cost for large-scale remediation |
| Alkali | suitable for pollutants that can react with ·OH | restricted by pH; equipment corrosion; precipitation of metal ions |
| UV | safe ; no secondary pollution | poor reusability; high energy consumption; high requirements for PS concentration |
| Carbonaceous materials | high degradation efficiency; cheap and easy to get | restricted by pH; poor stability |
| Electrochemical | economic and environmental friendly; controllable process; high degradation efficiency | pretreatment needed; electrode passivation |
3 SO4.-降解有机污染物
SO4.-降解有机污染物在机理上有三种途径:加成作用(addition)、夺氢作用(hydrogen abstraction)和直接电子转移(direct electron transfer)[22]。
加成作用主要发生在含碳碳双键或碳碳三键等不饱和键污染物的降解反应中,SO4.-成单的电子使不饱和键断裂,与SO4.-发生加成反应,达到降解污染物的目的[78]:
SO4.- +H2C=CHR→-OSO2OCH2-CHR·
夺氢作用主要发生在醇类、醚类和有机酸等的降解反应中,SO4.-直接夺取有机物羟基、醚键和羧基等官能团中的氢,并最终使有机物降解为CO2和H2O[78]:
SO4.- +RH→HSO4-+R·
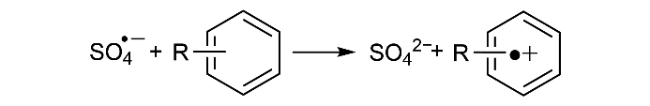
SO4.-对有机物的选择性反应顺序为:非芳香碳碳双键有机物>芳环上含π电子的有机物>含α-H的有机物>含非α-H的有机物[84]。因此不同污染物在使用SO4.-降解时,其降解机理和降解能力也是不同的。
3.1 POPs
POPs是一类毒性高、生物蓄积性强、难以自然降解并可远距离迁移的全球性有机污染物,主要包括有机氯农药(organochlorine pesticides,OCPs)、多氯联苯(polychlorinated biphenyls,PCBs)、多氯二苯并-对-二口恶英(polychlorinated dibenzo-para-dioxins,PCDDs)和多氯二苯并呋喃(polychlorinated dibenzofurans,PCDFs),以及近年来陆续新列入《关于持久性有机污染物的斯德哥尔摩公约》的多溴二苯醚(polybrominated diphenyl ethers,PBDEs)、全氟辛基磺酸(perfluorooctane sulfonic acid,PFOS)及其盐类、全氟辛基磺酰氟(perfluorooctane sulfonyl fluoride,PFOSF)、PFOA及其盐类和相关化合物等。此外,邻苯二甲酸酯(phthalic acid esters,PAEs,别名酞酸酯)[85]和PAHs[86]等迄今未被列入《斯德哥尔摩公约》的POPs清单,但不少学者将其视为POPs进行研究。
POPs因其难降解性,传统降解方法效率低且会产生有害副产物。SO4.-因其还原电位高被用于POPs的降解(表4 ),其中不含苯环的OCPs要比含苯环的PBDEs和PCBs等更易降解,如Khan等[87]用UV/Fe2+活化PMS降解林丹(lindane),40 min完全降解;而DDT等含苯环的OCPs和菲(phenanthrene)等PAHs降解率高的原因可能是苯环间不含联苯键、醚键等难断裂化学键。因此,苯环对SO4.-的氧化降解率有一定的影响。苯环数相同时,取代原子数越多越难被降解,如CB-28降解的表观速率常数kobs(0.49 h-1)低于CB-1(1.31 h-1)。
表4 SO4.-降解POPsTable 4 Degradation of POPs by SO4.- |
| Class | POPs | Conc. (μM) | PS | Conc. (μM) | Activator | Conc. | pH | T(℃) | t(h) | kobs() | Degradation rate | ref | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OCPs | endosulfan | 2.45 | PDS | 49 | 20∶1 | UV | — | 7 | 25 | 1.3 | — | 90% | 25 |
| lindane | 3.43 | PMS | 250 | 73∶1 | UV/Fe2+ | 0.05 mM | 3 | 25 | 0.6 | 5.05 | 98% | 87 | |
| DDT | 2.82 | PMS | 2000 | 710∶1 | Co2+ | 40 mM | 3~5 | 40 | 2 | 21.60 | 100% | 102 | |
| PCBs | CB-28 | 3.9 | PDS | 2000 | 513∶1 | V2O3 | 1.33 mM | 5.9 | 25 | 4 | 0.89 | 100% | 98 |
| CB-28 | 1.94 | PMS | 200 | 103∶1 | CuFe2O4 | 0.42 mM | 7 | 25 | 8 | 0.49 | 89% | 96 | |
| CB-1 | 20 | PMS | 400 | 20∶1 | Fe2+ | 0.2 mM | 3 | 25 | 4 | 1.31 | 90% | 103 | |
| PBDEs | BDE-47 | 3.1 | PDS | 71 400 | 23 032∶1 | nZVI | 1000 mg/L | 6~11 | 25 | 48 | — | 64% | 39 |
| BDE-209 | 5.2 | PDS | 100 000 | 19 183∶1 | BC-nZVI | 200 mM | 3 | 40 | 4 | 0.30 | 82% | 50 | |
| BDE-209 | 10.4 | PDS | 200 000 | 19 183∶1 | Fe2+ | 100 mM | 3 | 25 | 4 | 0.18 | 51% | 93 | |
| PFASs | PFOA | 120 | PDS | 12 000 | 100∶1 | AC-Fe | 1000 mg/L | 2.5 | 25 | 10 | 0.21 | 64% | 95 |
| PFOA | 0.5 | PDS | 20 000 | 40 000∶1 | heat | — | 2 | 85 | 8 | 0.83 | 98% | 99 | |
| PFOS | 0.92 | PDS | 84 000 | 91 304∶1 | heat | — | — | 90 | 70 | — | 0% | 100 | |
| PAHs | phenanthrene | 5.6 | PDS | 2000 | 357∶1 | V2O3 | 100 mg/L | 3~5 | 25 | 3.6 | 7.15 | 88% | 42 |
| Intermediate | p-chloroaniline | 500 | PDS | 2500 | 5∶1 | AC | 5000 mg/L | 3~9 | 20 | 0.5 | — | 98% | 104 |
SO4.-作为氧化剂及亲电子自由基,易与失电子能力强的基团反应[81],而PBDEs的取代基团Br比PCBs的Cl更易失电子[88],所以表4 中PCBs的降解速率比PBDEs快可能受醚键[89]等其他因素的影响。Dominguez等[90]热活化PDS降解林丹和联苯等多种氯代有机物,40 ℃时所有污染物在25~72 h内接近完全降解,但脱下的Cl-会在一定条件下与SO4.-反应生成降解能力弱的氯自由基[91],抑制氯代有机物的降解。溴代有机物PBDEs降解方面,近年的报道主要为nZVI或含铁复合物活化PS来降解[39,50,92],但降解率较低,有报道通过添加羟胺和抗坏血酸等还原剂及螯合剂可有限地提高降解率。如Peng等[93]用Fe2+活化PDS降解BDE-209,降解率仅51%,添加螯合剂柠檬酸盐和葡萄糖酸钠后,因其可与Fe2+结合,避免Fe2+与生成的SO4.-反应,降解率分别升至61%和72%;而添加抗坏血酸后,一方面作为活化剂活化PDS,使体系产生更多的SO4.-,另一方面也作为还原剂使Fe3+还原为Fe2+,再度参与反应,但抗坏血酸也能消耗SO4.-,所以其降解率仅增至61%。与Cl-一样,Br-也会与污染物竞争SO4.-,且Br-与SO4.-的反应速率更快(Cl-:2.7×108 M-1·S-1[91],Br-:3.5×109 M-1·S-1[27]),这可能也是溴代POPs比氯代POPs更难降解的一个原因。
PBDEs和PCBs降解率低的原因也可能是其取代基团均为吸电子基团,所以其降解反应主要通过加成反应进行,而不易通过速率更快的直接电子转移途径[88]。而精细化工产品的合成中间体对氯苯胺(p-chloroaniline)因其仅含两个取代基,且其中一个为给电子基团氨基,所以更易经直接电子转移途径降解,30 min降解率达98%,最终降解产物为CO2、H2O和HCl,并且该体系的pH(3~9)适应范围广(表4 )。
超氧自由基(O2.-)对降解反应也有一定的影响,如Qin等[96]用CuFe2O4活化PMS降解CB-28,经电子顺磁共振(electron paramagnetic resonance,EPR)检测证实了O2.-的存在,通过添加O2.-猝灭剂对苯醌(p-benzoquinone,BQ)和超氧化物歧化酶(superoxide dismutase,SOD)使降解率从89%分别降至58%和19%,其中添加SOD后降解率更低的原因是其将O2.-分解生成H2O2,H2O2与PDS竞争消耗Fe2+,降低SO4.-的产生量,而BQ则直接将O2.-猝灭。在增加体系溶解氧量时,降解率增至98%,也证实O2.-有一定的降解作用。
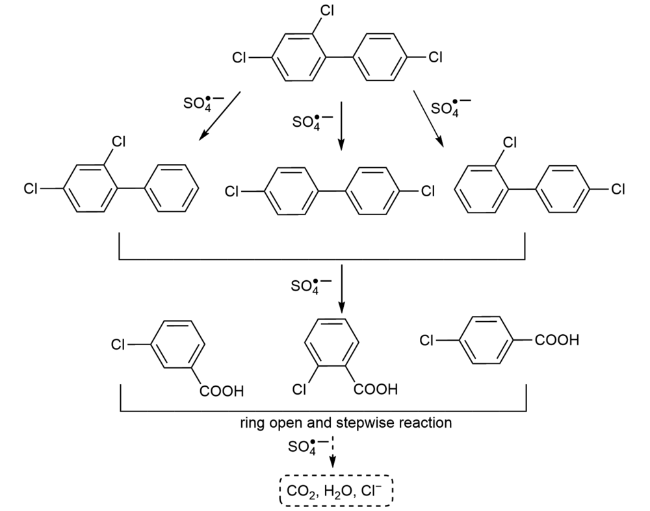
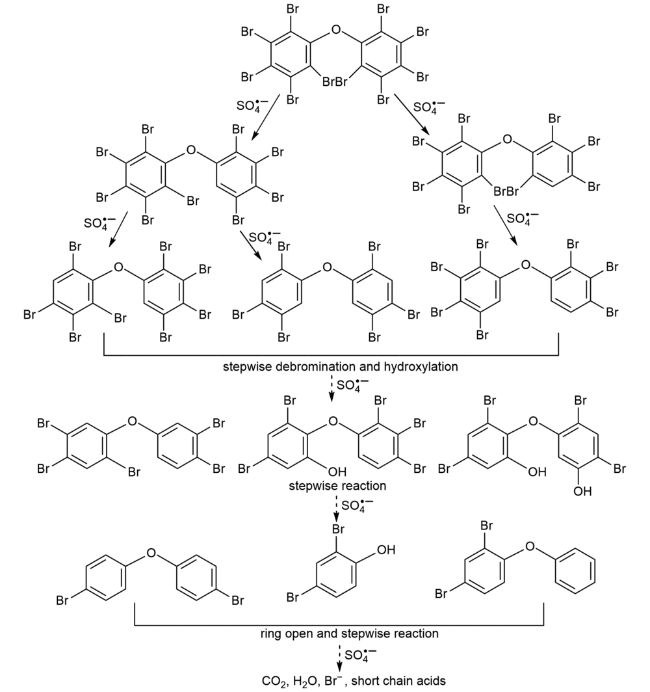
大量研究表明SO4.-降解POPs的中间产物后续大多可被降解并矿化。OCPs作为较易降解的POPs,其矿化程度也较高。Khan等[87]用UV/Fe2+活化PMS降解林丹,180 min时其脱氯率与TOC去除率分别为96.4%和92.2%。硫丹降解的主要副产物为硫丹醚,与直接UV光解相比,Shah等[25]发现UV活化PS产生的SO4.-能更有效地去除硫丹醚,从而避免其对环境的不利影响。DDT最终降解产物与PCBs相同[97],都被矿化为CO2、H2O、Cl-,不同的是DDT降解过程中苯环上羧基化和羟基化两种加成反应都会发生,而PCBs降解时苯环上以羧基化加成为主(图3 ,据Qin等[96]、Fang等[98]报道的降解途径总结得出),而PBDEs降解时苯环上为羟基化加成[50],PBDEs先脱溴,后在自由基作用下发生醚键断裂[41]并开环完成降解(图4 ,据Peng等[41]、Li等[50]、Peng等[65]报道的降解途径总结得出)。Li等[50]用BC-nZVI活化PDS降解BDE-209,经GC-MS检测得出BDE-209在转化为低溴中间体后,最终被矿化为CO2、H2O、Br-以及一些短链酸。Huang等[31]用机械化学活化PDS降解BDE-209的结果与前者不同,BDE-209直接与SO4.-作用,产物为CO2和Br2,降解率、脱溴率均接近100%,经HPLC检测不存在低溴中间产物。由此可见,不同活化方法的降解途径可能是不同的,应根据污染物的性质选择降解效果好的活化方法。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Chen等[42]用V2O3活化PDS降解菲,菲经直接电子转移途径降解,LC-MS检测显示菲经开环转化为短链酸后,最终被降解为CO2和H2O,可见PAHs也可用SO4.-降解。PFOA通过依次脱去—CF2降解,因此会产生有害的短链全氟烷基物质[94]。Bruton等[99]用热活化PDS降解PFOA,85 ℃下降解率达98%,但其中34%未完全脱氟,以C4~C7的短链全氟烷基物质存在于溶液中,因此需要重复投加PDS才能完全去除PFOA及其副产物。90 ℃下热活化PDS降解另一种常见的全氟烷基物质PFOS则不发生降解[100],可能是由于C—F还原电位(3.6 eV)高于SO4.-,所以SO4.-先和C—S键反应,之后再依次脱氟,所以更难降解,这也是在更高温度(350 ℃)及压力(275.8 kPa)下PFOS降解率只有22%的原因[101]。而Wang等[32]以机械化学活化PDS降解PFOA,虽然也产生短链全氟烷基物质,但经GC-MS检测显示它们最终被完全降解,PFOA的脱氟率达到100%,完全矿化为CO2以及游离的F-,但该方法对PFOS也仅有56%的降解率及9.7%的脱氟率。
3.2 PPCPs
目前,水体中PPCPs的降解技术主要有生物接触氧化、臭氧氧化、光催化氧化和膜分离技术等[106],这些方法对PPCPs的选择性较低,而SO4.-还原电位高,选择性好,适用于含药物废水的深度降解[8],如CBZ的降解[108]。Lian等[22]用SO4.-降解15种常见PPCPs的混合液,8种PPCPs降解率超60%,且其中4种近100%,而传统H2O2产生的·OH降解15种PPCPs时,最高降解率仅65%左右。Mahdi Ahmed[109]在超纯水与废水(包含活性污泥及Cl-、NO3-、HCO3-、Na+等离子)中分别用Co2+/PMS(SO4.-)和芬顿体系(·OH)降解SMX和双氯芬酸钠(diclofenac,DCF)。超纯水条件下,SO4.-和·OH降解率相似,30 min SMX和DCF都达到100%的降解率;废水条件下,由于杂质的影响,SO4.-对SMX和DCF的降解率降至81%和71%,而·OH则大幅降至18%和26%,·OH因其选择性差而更多地被其他物质消耗,降解效果远不及SO4.-。但是,SO4.-降解PPCPs也会受废水中腐殖酸等有机物及Cl-和HCO3-等阴离子的干扰,所以降解前应先去除杂质离子[110]。
表5 SO4.-降解抗生素Table 5 Degradation of antibiotics by SO4.- |
| Class | Antibiotic | Conc. (mM) | PS | Conc. (mM) | MPS∶ Pollutant | Activator | Conc. | pH | T(℃) | t(h) | kobs(h-1) | Degradation rate | ref |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fluoroquinolones | ciprofloxacin | 0.04 | PDS | 0.5 | 12.5∶1 | UV254 | — | 6.5 | — | 0.16 | — | 70% | 67 |
| norfloxacin | 0.03 | PDS | 4.4 | 147∶1 | BC | 800 mg/L | 6.5 | 25 | 5 | 0.57 | ~100% | 56 | |
| Sulfonamides | sulfathiazole | 0.10 | PDS | 5 | 50∶1 | Cu2+ | 0.5 mM | 7 | 25 | 72 | 0.04 | 95% | 114 |
| sulfamethazine | 0.18 | PMS | 1 | 5.6∶1 | CuCo2O4 | 100 mg/L | 5 | 25 | 0.5 | 8.46 | 95% | 111 | |
| Tetracyclines | tetracycline | 0.34 | PDS | 11.1 | 32.9∶1 | Fe | 3500 mg/L | 3.6 | — | 3 | — | 81.6% | 115 |
| β-Lactams | cephalosporin | 0.10 | PDS | 1.1 | 11∶1 | Cu2+ | 0.05 mM | 7 | — | 2 | — | ~100% | 26 |
SO4.-降解PPCPs的中间产物后续也能被氧化降解。Zhang等[36]用纳米Co3MnFeO6活化PMS降解药物CBZ,30 min降解率为100%,结果显示CBZ被SO4.-降解为分子量更小的有机物后最终矿化为CO2和H2O。Pi等[57]制备氧化BC负载Fe3O4活化PDS降解四环素,1 h降解率92.3%,而矿化度仅40%,但中间产物最终也被分解为CO2和H2O。磺胺二甲嘧啶[111]、诺氟沙星[38]以及头孢菌素(cephalosporin)[113]的降解也有相似的结果。由此可见,基于SO4.-的AOPs降解PPCPs,在SO4.-足量且时间足够长的理想情况下,中间产物最终都能被矿化,基本不产生二次污染,是一种环境友好的方法。但在实际工程应用条件下,因成本、活化剂及时间的问题,很难达到使其完全矿化的条件。此外,降解反应中未消耗的PS及过量的自由基等残留物[31]也可能对环境有一定的影响,所以其工程应用还需要进一步的研究。
3.3 有机染料
有机染料广泛用于纺织和造纸等行业,印染废水毒性强、色度深、难以生化降解,是处理难度高的工业废水之一。光催化氧化、生物处理和吸附等传统处理技术有流程慢、易造成二次污染和成本高等缺点[116]。
近年来,基于SO4.-的AOPs在有机染料废水处理上的应用逐渐受到关注。Jeon等[117]用Fe2+活化PDS降解橙黄,降解率达100%。为降低过量Fe2+消耗SO4.-,引入了Fe阳极电流法,不仅缓慢增加Fe2+,阴极上Fe3+也被还原为Fe2+并进一步参与反应。而Li等[52]用还原氧化石墨烯(reduced graphene oxide,rGO)负载Co-Mn层状双金属氢氧化物(layered double hydroxide,LDH)活化PMS降解酸性橙,石墨烯层不但能通过改善金属氧化物的分散性而减少金属的浸出,增强两组分间的协同作用,而且也有效阻止了氧化物粒子间的团聚,使其更多地与PMS接触,从而产生更多的SO4.-,达到完全降解。Zhao等[118]用SO4.-降解酸性橙、亚甲基蓝(methylene blue)、罗丹明B(rhodamine B)、甲基橙(methyl orange)和甲基玫瑰苯胺氯化物等几种典型有机染料混合液,除甲基玫瑰苯胺氯化物降解率仅90%外,其余均达到100%且pH具有广适性(3~10)。
由于废水环境的特殊性,许多杂质离子会影响降解率。Ji等[121]发现水中NO2-在SO4.-降解氯酚的过程中,不仅消耗SO4.-使降解率下降,生成的二氧化氮自由基也参与反应生成毒性更大的副产物氯硝基苯酚。
与PPCPs相似,有机染料之间结构差异也较大,导致其降解路径各异。Hou等[45]用GC-MS检测分析SO4.-降解酸性橙的反应中间体及产物,结果显示SO4.-通过加成作用先与偶氮键反应后,再攻击C=N不饱和键,之后形成一系列中间体,这些中间体由于均不含联苯键或醚键等难断裂化学键,最终易开环转化为CO2、H2O及一些小分子有机物。而Yi等[122]通过HPLC-MS检测SO4.-降解罗丹明B的反应中间体及产物,SO4.-先通过夺氢途径脱去罗丹明B最外侧的乙基,然后形成的芳香环中间体在SO4.-作用下开环分解为小分子脂肪酸和短链烯烃,最终被矿化为CO2、H2O和小分子有机物(醇、有机酸和胺)。由此可见,SO4.-降解有机染料的最终产物除CO2和H2O外,还包括有机酸和醇类等小分子有机物,但对环境无害。
表6 SO4.-降解有机染料Table 6 Degradation of organic dyes by SO4.- |
| Organic dye | Conc. (mM) | PS | Conc. (mM) | MPS/Pollutant | Activator | Conc. | pH | T(℃) | t(min) | kobs(h-1) | Degradation rate | ref | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Acid orange 7 | 0.2 | PDS | 4 | 20∶1 | CoMn2O4/rGO | 50 mg/L | 7 | 25 | 20 | — | 100% | 52 | ||||
| Acid orange 7 | 0.06 | PMS | 0.55 | 9.7∶1 | Mn-Fe/LDH | 200 mg/L | 5~9 | 25 | 30 | 8.28 | 98% | 45 | ||||
| Acid orange G | 0.11 | PMS | 0.28 | 2.5∶1 | Co-Mn/LDH | 25 mg/L | 3~10 | 25 | 4 | 50.40 | 100% | 118 | ||||
| Methylene blue | 0.16 | 1.8∶1 | — | 100% | ||||||||||||
| Rhodamine B | 0.10 | 2.7∶1 | — | 100% | ||||||||||||
| Methyl orange | 0.15 | 1.8∶1 | — | 100% | ||||||||||||
| p-Chloronitrobenzene | 2.7 | PDS | 200 | 74.1∶1 | ZVI | 1.0 mmol/g | 3~7 | 25 | 480 | — | 90% | 46 | ||||
4 结论及展望
基于SO4.-的AOPs降解新型有机污染物,在降解率和适应性等方面均优于·OH,研究和应用前景广阔。虽然SO4.-降解有机污染物的报道较多,但目前仍处在实验研究阶段,其工程应用还有待深入研究。对SO4.-降解体系的研究应关注过硫酸盐的充分活化、降低成本和减少副产物等方面。
(1)根据不同污染物的结构和理化性质选择最佳的活化方式,通过改进现有活化技术或联用多种活化方式,提高污染物的降解率和矿化度。
(2)开发低成本、高效且稳定的新型天然活化剂,降低处理成本并减少二次污染。
(3)深入研究多种混合物体系中SO4.-降解反应的机制,并尽可能降低杂质离子对降解反应的干扰,开展废水和土壤等实际环境介质中SO4.-降解新型有机污染物的研究,以进一步推广应用该技术。
(4)在实际应用中受杂质等的干扰,SO4.-降解某些有机污染物时,可能会产生更难降解甚至更大毒性的中间体、副产物及残留物,需对其进行监测分析并设法消除其不良影响。