
文章编号: 190905
文献标识码: A
液质联用中接口离子化新技术
收稿日期:2019-09-02
修回日期:2019-12-25
网络出版日期:2020-02-20
基金资助
国家自然科学基金项目(21672249)
版权
New Ionization Technology for Interface of Liquid Chromatography-Mass Spectrometry
Received:2 Sept. 2019
Revised:25 Dec. 2019
Online:20 Feb. 2020
Fund
National Natural Science Foundation of China(21672249)
Copyright
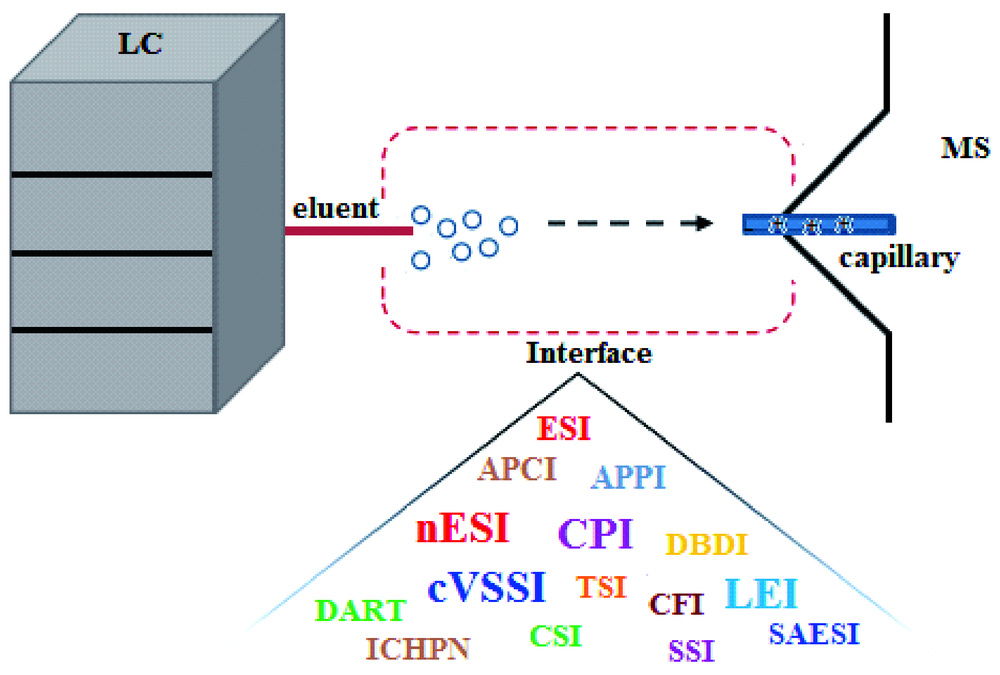
液相色谱-质谱联用(简称液质联用,LC-MS)将色谱的高分离效能与质谱强大的结构测定功能结合,不仅实现了对复杂混合物更准确的定性定量分析,而且简化了样品的前处理过程,使样品分析更简便,在药物分析、食品与环境分析以及生物样品检测等众多领域得到了广泛的应用。作为LC-MS的核心组成部分,液质接口的作用是将LC的液体引入,发生电离,并将生成的离子传输进MS。因此,接口离子化技术的改进直接影响了LC-MS的发展和应用。为了获得更高的灵敏度和更广泛的适用性,研究人员一直致力于离子化技术的研究,以促进分析物的解吸,提高其电离和传输效率,减少基质效应的干扰。本文针对近年来LC-MS接口离子化技术的改进和发展,从离子化原理出发,对接口离子源的构造、影响电离的因素、以及相关的应用进行综述,探讨其优缺点,并对LC-MS接口离子化技术的发展趋势进行了展望。
田甜 , 张芳 , 张曙盛 , 冯陈国 , 苏越 , 林国强 . 液质联用中接口离子化新技术[J]. 化学进展, 2020 , 32(5) : 604 -616 . DOI: 10.7536/PC190905
Tian Tian , Fang Zhang , Shusheng Zhang , Chenguo Feng , Yue Su , Guoqiang Lin . New Ionization Technology for Interface of Liquid Chromatography-Mass Spectrometry[J]. Progress in Chemistry, 2020 , 32(5) : 604 -616 . DOI: 10.7536/PC190905
Liquid chromatography-mass spectrometry(LC-MS) combines the high separation efficiency of chromatography with the powerful structural determination of mass spectrometry, which not only enables more accurate analysis for the compounds, but also simplifies the pre-treatment of samples and makes the analysis more convenient. LC-MS, as an important tool for the qualitative and quantitative analysis of organic compounds, has been widely used in various fields such as pharmaceutical analysis, food and environmental monitoring, biological and medical research etc. As the key component of LC-MS, the role of the interface is to introduce and ionize the fractions from LC, and transfer the generated ions into the MS. Therefore, the improvement of ionization technology for the interface directly affects the advance and application of LC-MS. In order to obtain a higher sensitivity and a wider range of applicability, researchers have focused on the ionization technology to promote the desorption of chemicals, improve their ionization and transport efficiency, and reduce the interference of matrix effect. In this work, the development of traditional ionization technologies and the novel technologies reported for the interface of LC-MS in recent years are reviewed, including the ionization principle, interface construction, influencing factors, and the related applications. Their characteristics, advantages and disadvantages are discussed in details. Finally, the trend of ionization technology in development for the interface of LC-MS is prospected.
1 Introduction
2 Interface ionization technologies in LC-MS
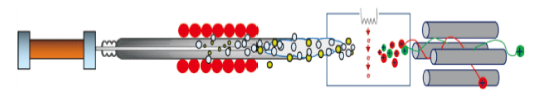
2.1 Electrospray ionization-related techniques
2.2 Plasma-based ionization
2.3 Inlet ionization
2.4 Carbonfiber ionization
2.5 Capillary photoionization
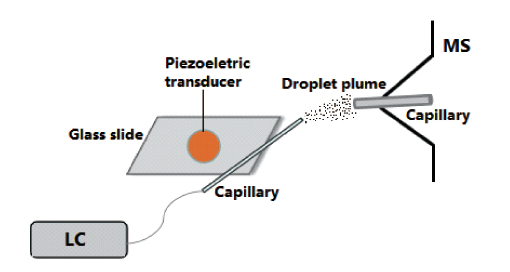
2.6 Capillary vibrating sharp-edge spray ionization
2.7 Liquid electron ionization
2.8 Other ionization techniques
3 Conclusion and outlook
Key words: LC-MS ; interface ; ionization
| [1] |
Takats Z, Wiseman J M, Gologan B, Cooks R G. Science, 2004,306(5695):471. https://www.sciencemag.org/lookup/doi/10.1126/science.1104404
|
| [2] |
Hiraoka K, Nishidate K, Mori K, Asakawa D, Suzuki S. Rapid Commun. Mass Spectrom., 2007,21(18):3139. http://doi.wiley.com/10.1002/%28ISSN%291097-0231
|
| [3] |
Wilm M S, Mann M. International J. Mass Spectrom., 1994,136(2/3):167. https://linkinghub.elsevier.com/retrieve/pii/0168117694040249
|
| [4] |
Zhang J T, Wang H Y, Zhu W, Cai T T, Guo Y L. Anal. Chem., 2014,86(18):8937. https://pubs.acs.org/doi/10.1021/ac502656a
DOI: 10.1021/ac502656a |
| [5] |
Wilm M, Mann M. Anal. Chem., 1996,68(1):1.
|
| [6] |
Schupke H, Hempel R, Eckardt R, Kronbach T. J. Mass Spectrom., 1996,31(12):1371. http://doi.wiley.com/10.1002/%28ISSN%291096-9888
|
| [7] |
Mavroudakis L, Mavrakis E, Kouvarakis A, Pergantis S A. Rapid Commun. Mass Spectrom., 2017,31(11):911. http://doi.wiley.com/10.1002/rcm.7866
DOI: 10.1002/rcm.7866 |
| [8] |
Wang T W, Nam P K S, Shi H L, Ma Y F. J. Chromatogra. Sci., 2007,45(4):200. https://academic.oup.com/chromsci/article-lookup/doi/10.1093/chromsci/45.4.200
|
| [9] |
Benijts T, Gunther W, Lambert W, De Leenheer A. Rapid Commun. Mass Spectrom., 2003,17(16):1866. http://doi.wiley.com/10.1002/rcm.1131
DOI: 10.1002/rcm.1131 |
| [10] |
Huang Y, Zhang Q, Liu Y, Jiang B, Xie J, Gong T, Jia B, Liu X, Yao J, Cao W, Shen H, Yang P. Talanta, 2020,207:120340. https://linkinghub.elsevier.com/retrieve/pii/S0039914019309737
|
| [11] |
Rahman M M, Wu D, Chingin K, Xu W, Chen H. Talanta, 2019,202:59. https://linkinghub.elsevier.com/retrieve/pii/S0039914019304412
|
| [12] |
Berg A, Svobodova1 H, Dewberry A, 王勇为(Wang Y W), Valaskovic G. 中国化学会第二届全国质谱分析学术报告会( The 2nd National Conference on Mass Spectrometry Analysis of Chinese Chemical Society), 2015.
|
| [13] |
Keating J E, Glish G L. Anal. Chem., 2018,90(15):9117. https://pubs.acs.org/doi/10.1021/acs.analchem.8b01528
|
| [14] |
张骏婷(Zhang J T). 中国科学院上海有机化学研究所博士论文( Doctorial Dissertation of Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences), 2016.
|
| [15] |
Cody R B, Laramee J A, Durst H D. Anal. Chem., 2005,77(8):2297. https://pubs.acs.org/doi/10.1021/ac050162j
DOI: 10.1021/ac050162j |
| [16] |
Na N, Zhao M X, Zhang S C, Yang C D, Zhang X R. J. Am. Soc. Mass Spectrome., 2007,18(10):1859. https://pubs.acs.org/doi/10.1021/jasms.8b02797
|
| [17] |
Harper J D, Charipar N A, Mulligan C C, Zhang X R, Cooks R G, Ouyang Z. Anal. Chem., 2008,80(23):9097. https://pubs.acs.org/doi/10.1021/ac801641a
DOI: 10.1021/ac801641a |
| [18] |
Badal S P, Michalak S D, Chan G C, You Y, Shelley J T. Anal. Chem., 2016,88(7):3494. https://pubs.acs.org/doi/10.1021/acs.analchem.5b03434
|
| [19] |
Ratcliffe L V, Rutten F J M, Barrett D A, Whitmore T, Seymour D, Greenwood C, Aranda-Gonzalvo Y, Robinson S, McCoustra M. Anal. Chem., 2007,79(16):6094. https://pubs.acs.org/doi/10.1021/ac070109q
DOI: 10.1021/ac070109q |
| [20] |
Gross J H. Anal. Bioanal. Chem., 2014,406(1):63. http://link.springer.com/10.1007/s00216-013-7316-0
|
| [21] |
Chang C L, Xu G G, Bai Y, Zhang C S, Li X J, Li M, Liu Y, Liu H W. Anal. Chem., 2013,85(1):170. https://pubs.acs.org/doi/10.1021/ac303450v
DOI: 10.1021/ac303450v |
| [22] |
Chang C L, Zhou Z G, Yang Y Y, Han Y H, Bai Y, Zhao M P, Liu H W. Electrophoresis, 2012,33(22):3387. http://dx.doi.org/10.1002/elps.201200122
Normal phase chiral LC (NPLC) has been proved to be powerful and efficient for chiral separation. However, the combination of NPLC with ESI or atmospheric pressure chemical ionization MS is restricted by the poor ionization efficiency and thermal fragmentations of analytes to some extent. Direct analysis in real time MS (DART-MS) is an ambient ionization technique that shows high ionization efficiency of the analytes in the normal phase mobile phase. In this work, we coupled chiral NPLC to DART-MS for the chiral qualitative and quantitative analysis of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol and jasmonic acid enantiomers. Satisfactory results for the enantiomers of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol operating in the positive mode were obtained in terms of linearity (2.5250 mu g/mL, R-2, 0.999-1.000) and repeatability (25 mu g/mL, RSDs, 4.7-5.6%). Moreover, chiral NPLC-DART-MS resulted in the simultaneous chiral separation and detection of jasmonic acid enantiomers, which are very difficult to be analyzed by NPLC-ESI-MS and NPLC-APCI-MS. Compared with the coupled techniques of NPLC-ESI-MS and NPLC-APCI-MS, NPLC-DART-MS showed advantages in increasing the ionization efficiency and reducing the in-source thermal fragmentation of analytes. |
| [23] |
Hayen H, Michels A, Franzke J. Anal. Chem., 2009,81(24):10239. https://pubs.acs.org/doi/10.1021/ac902176k
DOI: 10.1021/ac902176k |
| [24] |
Gilbert-Lopez B, Lara-Ortega F J, Robles-Molina J, Brandt S, Schutz A, Moreno-Gonzalez D, Garcia-Reyes J F, Molina-Diaz A, Franzke J. Anal. Bioanal. Chem., 2019,41(19):4785.
|
| [25] |
Gilbert-Lopez B, Garcia-Reyes J F, Meyer C, Michels A, Franzke J, Molina-Diaz A, Hayen H. Analyst, 2012,137(22):5403. http://dx.doi.org/10.1039/c2an35705d
DOI: 10.1039/c2an35705d |
| [26] |
Covey T R, Thomson B A, Schneider B B. Mass Spectrom. Rev., 2009,28(6):870. http://doi.wiley.com/10.1002/mas.v28%3A6
DOI: 10.1002/mas.v28:6 |
| [27] |
Page J S, Kelly R T, Tang K, Smith R D. J. Am. Soc. Mass Spectrom., 2007,18(9):1582. https://pubs.acs.org/doi/10.1021/jasms.8b03007
|
| [28] |
Kelly R T, Tolmachev A V, Page J S, Tang K, Smith R D. Mass Spectrom. Rev., 2010,29(2):294.
|
| [29] |
Trimpin S, Inutan E D, Herath T N, McEwen C N. Mol. Cell Proteomics, 2010,9(2):362. http://www.mcponline.org/lookup/doi/10.1074/mcp.M900527-MCP200
|
| [30] |
Trimpin S, Inutan E D, Herath T N, McEwen C N. Anal. Chem., 2010,82(1):11. https://pubs.acs.org/doi/10.1021/ac902066s
DOI: 10.1021/ac902066s |
| [31] |
Pagnotti V S, Inutan E D, Marshall D D, McEwen C N, Trimpin S. Anal. Chem., 2011,83(20):7591. https://pubs.acs.org/doi/10.1021/ac201982r
DOI: 10.1021/ac201982r |
| [32] |
Vestal M L. Mass Spectrom. Rev., 1983,2(4):447. http://doi.wiley.com/10.1002/%28ISSN%291098-2787
|
| [33] |
Pagnotti V S, Chubatyi N D, McEwen C N. Anal. Chem., 2011,83(11):3981. https://pubs.acs.org/doi/10.1021/ac200556z
DOI: 10.1021/ac200556z |
| [34] |
Pagnotti V S, Chakrabarty S, Harron A F, McEwen C N. Anal. Chem., 2012,84(15):6828. https://pubs.acs.org/doi/10.1021/ac3014115
DOI: 10.1021/ac3014115 |
| [35] |
Chubatyi N D, Pagnotti V S, Bentzley C M, McEwen C N. Rapid Commun. Mass Spectrom., 2012,26(8):887. http://doi.wiley.com/10.1002/rcm.6179
DOI: 10.1002/rcm.6179 |
| [36] |
Wang B X, Inutan E D, Trimpin S. J. Am. Soc. Mass Spectrom., 2012,23(3):442. https://pubs.acs.org/doi/10.1021/jasms.8b04246
|
| [37] |
Fenner M A, Chakrabarty S, Wang B, Pagnotti V S, Hoang K, Trimpin S, McEwen C N. Anal. Chem., 2017,89(9):4798. https://pubs.acs.org/doi/10.1021/acs.analchem.6b05172
|
| [38] |
Liu J, Ro K W, Busman M, Knapp D R. Anal. Chem., 2004,76(13):3599. https://pubs.acs.org/doi/10.1021/ac030419i
DOI: 10.1021/ac030419i |
| [39] |
Ro K W, Liu H, Busman M, Knapp D R. J. Chromatogra. A, 2004,1047(1):49. https://linkinghub.elsevier.com/retrieve/pii/S0021967304010878
|
| [40] |
Wu M X, Wang H Y, Zhang J T, Guo Y L. Anal. Chem., 2016,88(19):9547. https://pubs.acs.org/doi/10.1021/acs.analchem.6b02166
|
| [41] |
Wu M L, Chen T Y, Chen Y C, Chen Y C. Anal. Chem., 2017,89(24):13458. https://pubs.acs.org/doi/10.1021/acs.analchem.7b03736
|
| [42] |
吴梦茜(Wu M X). 中国科学院上海有机化学研究所博士论文( Doctorial Dissertation of Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences), 2017.
|
| [43] |
Robb D B, Covey T R, Bruins A P. Anal. Chem., 2000,72(15):3653. https://pubs.acs.org/doi/10.1021/ac0001636
DOI: 10.1021/ac0001636 |
| [44] |
Kersten H, Derpmann V, Barnes I, Brockmann K J, O’Brien R, Benter T. J. Am. Soc. Mass Spectrom., 2011,22(11):2070. https://pubs.acs.org/doi/10.1021/jasms.8b03917
|
| [45] |
Haapala M, Suominen T, Kostiainen R. Anal. Chem., 2013,85(12):5715. https://pubs.acs.org/doi/10.1021/ac4002673
DOI: 10.1021/ac4002673 |
| [46] |
Poho P, Vaikkinen A, Haapala M, Kylli P, Kostiainen R. Analyst, 2019,144(9):2867. http://xlink.rsc.org/?DOI=C9AN00258H
DOI: 10.1039/C9AN00258H |
| [47] |
Li X J, Attanayake K, Valentine S J, Li P. Rapid Commun. Mass Spectrom., 2018.
|
| [48] |
Ranganathan N, Li C, Suder T, Karanji A K, Li X J, He Z Y, Valentine S J, Li P. J. Am. Soc. Mass Spectrom., 2019,30(5):824. https://doi.org/10.1007/s13361-019-02147-0
|
| [49] |
Heron S R, Wilson R, Shaffer S A, Goodlett D R, Cooper J M. Anal. Chem., 2010,82(10):3985. https://pubs.acs.org/doi/10.1021/ac100372c
DOI: 10.1021/ac100372c |
| [50] |
Amirav A, Granot O. J. Am. Soc. Mass Spectrom., 2000,11(6):587. https://pubs.acs.org/doi/10.1021/jasms.8b01488
|
| [51] |
Cappiello A, Balogh M, Famiglini G, Mangani F, Palma P. Anal. Chem., 2000,72(16):3841. https://pubs.acs.org/doi/10.1021/ac991493x
DOI: 10.1021/ac991493x |
| [52] |
Cappiello A, Famiglini G, Mangani F, Palma P. Mass Spectrom. Rev., 2001,20(2):88. http://doi.wiley.com/10.1002/%28ISSN%291098-2787
|
| [53] |
Tomasini D, Cacciola F, Rigano F, Sciarrone D, Donato P, Beccaria M, Caramao E B, Dugo P, Mondello L. Anal. Chem., 2014,86(22):11255. https://pubs.acs.org/doi/10.1021/ac5038957
DOI: 10.1021/ac5038957 |
| [54] |
Termopoli V, Famiglini G, Palma P, Piergiovanni M, Cappiello A. Anal. Chem., 2017,89(3):2049. https://pubs.acs.org/doi/10.1021/acs.analchem.6b04646
|
| [55] |
Cappiello A, Famiglini G, Palma P, Pierini E, Trufelli H, Maggi C, Manfra L, Mannozzi M. Chemosphere, 2007,69(4):554. https://linkinghub.elsevier.com/retrieve/pii/S0045653507003943
|
| [56] |
Cappiello A, Famiglini G, Palma P, Pierini E, Termopoli V, Trufelli H. Mass Spectrom. Rev., 2011,30(6):1242. http://dx.doi.org/10.1002/mas.20329
DOI: 10.1002/mas.20329 This review article will give an up-to-date and exhaustive overview on the efficient use of electron ionization (EI) to couple liquid chromatography and mass spectrometry (LC-MS) with an innovative interface called Direct-EI. EI is based on the gas-phase ionization of the analytes, and it is suitable for many applications in a wide range of LC-amenable compounds. In addition, thanks to its operating principles, it prevents unwelcome matrix effects (ME). In fact, although atmospheric pressure ionization (API) methodologies have boosted the use of LC-MS, the related analytical methods are sometime affected by inaccurate quantitative results, due to unavoidable and unpredictable ME. In addition, API's soft ionization spectra always demand for costly and complex tandem mass spectrometry (MS/MS) instruments, which are essential to acquire an "information-rich" spectrum and to obtain accurate quantitative information. In EI a one-stage analyzer is sufficient for a qualitative investigation and MS/MS detection is only used to improve sensitivity and to cut chemical noise. The technology illustrated here provides a robust and straightforward access to classical, well-characterized EI data for a variety of LC applications, and readily interpretable spectra for a wide range of areas of research. The Direct-EI interface can represent the basis for a forthcoming universal LC-MS detector for small molecules. (C) 2011 Wiley Periodicals, Inc., Mass Spec Rev 30:1242-1255, 2011 |
| [57] |
Palma P, Famiglini G, Trufelli H, Pierini E, Termopoli V, Cappiello A. Anal. Bioanal. Chem., 2011,399(8):2683. http://link.springer.com/10.1007/s00216-010-4637-0
|
| [58] |
Cappiello A, Famiglini G, Termopoli V, Trufelli H, Zazzeroni R, Jacquoilleot S, Radici L, Saib O. Anal. Chem., 2011,83(22):8537. https://pubs.acs.org/doi/10.1021/ac201839x
DOI: 10.1021/ac201839x |
| [59] |
Trufelli H, Famiglini G, Termopoli V, Cappiello A. Anal. Bioanal. Chem., 2011,400(9):2933. http://link.springer.com/10.1007/s00216-011-4955-x
|
| [60] |
Cappiello A, Famiglini G, Palma P, Termopoli V, Trufelli H. J. Chromatogr. A, 2012,1255:286. http://dx.doi.org/10.1016/j.chroma.2011.12.068
DOI: 10.1016/j.chroma.2011.12.068 One of the crucial tasks of pharmaceutical industry is to quantify the potential genotoxic impurities (PGIs) coming from the process of drug production. The European Medicines Agency (EMEA) imposes analytical testing limits in the order of mu g/g, depending on drug dosage and exposure period, that means the need of a sensitive and selective method of analysis. Liquid chromatography coupled to electrospray ionization mass spectrometry (LC-ESI-MS) has been demonstrated as the most versatile approach to detect PGIs in complex matrices. However, time consuming derivatization processes are needed to enhance sensitivity and selectivity, and to overcome matrix effects (ME) that may arise from active pharmaceutical ingredients (APIs) or excipients. We propose the use of the Direct-EI LC-MS as an alternative approach to detect and quantify PGIs in drug formulations. The Direct-EI LC-MS interface is based on electron ionization (El) which is well suited for the detection of low molecular weight compounds of different polarity, without derivatization and with no sign of ME. The method has been successfully applied to the detection of PGIs belonging to the class of alkylation agents. Calibration experiments show satisfactory linearity and precision data. Recoveries in low enriched samples spanned from 55 to 82%, and were not affected by ME. The method limits of detection (LODs), varying from 0.13 to 1.5 mu g/g, were satisfactory for the quantitation of the target PGIs at the level required by regulatory agencies. (C) 2011 Elsevier B.V. |
| [61] |
Cappiello A, Tirillini B, Famiglini G, Trufelli H, Termopoli V, Flender C. Phytochem. Anal., 2012,23(3):191. http://doi.wiley.com/10.1002/pca.v23.3
DOI: 10.1002/pca.v23.3 |
| [62] |
Famiglini G, Termopoli V, Palma P, Capriotti F, Cappiello A. Electrophoresis, 2014,35(9):1339. http://onlinelibrary.wiley.com/doi/10.1002/elps.201300462/abstract
An LC-MS method for the analysis of personal care and household products without sample preparation is presented. The method takes advantage of the Direct-electron ionization (EI) LC-MS interface for the quantitation of principal components, as well as for the identification of unknown or undeclared ingredients. The technique has proven its inertness toward matrix effects and the electron ionization allows quantitation and library identification. Commercially available products (shower gel, perfume, and hand cream) were diluted with methanol and injected directly into a nano-LC column. Limonene, linalool, and citral were selected as target compounds because of their use as fragrances in toiletry and detergent products. These and all other fragrances are commonly determined with GC-MS analysis, prior to sample cleanup, a procedure that can lead to analytes loss. The selected compounds are not detected with ESI because of their poor or very low response. Figures of merit and validation studies were executed and special attention was devoted to matrix-effects evaluation, because a sample preparation procedure is not involved. No matrix effects were observed, and the repeatability was excellent even after several weeks of operation. Products composition was investigated in full scan mode to determine the presence of unknown or not listed ingredients. |
| [63] |
Termopoli V, Famiglini G, Palma P, Piergiovanni M, Rocio-Bautista P, Ottaviani M F, Cappiello A, Saeed M, Perry S. J. Chromatogr. A, 2019,1591:120. https://linkinghub.elsevier.com/retrieve/pii/S0021967319300494
|
| [64] |
Popov R S, Ivanchina N V, Kicha A A, Malyarenko T V, Dmitrenok P S. J. Am. Soc. Mass Spectrom., 2019,30(5):743. https://doi.org/10.1007/s13361-019-02136-3
|
| [65] |
Cheng H Y, Zhang W W, Wang Y C, Liu J H. J. Chromatogr. A, 2018,1575:59. https://linkinghub.elsevier.com/retrieve/pii/S0021967318311749
|
| [66] |
Dahal U P, Jones J P, Davis J A, Rock D A. Drug MeTab. Dispos., 2011,39(12):2355. http://dx.doi.org/10.1124/dmd.111.040865
Identification and quantification of the metabolites of drugs and drug candidates are routinely performed using liquid chromatography-mass spectrometry (LC-MS). The best practice is to generate a standard curve with the metabolite versus the internal standard. However, to avoid the difficulties in metabolite synthesis, standard curves are sometimes prepared using the substrate, assuming that the signal for substrate and the metabolite will be equivalent. We have tested the errors associated with this assumption using a series of very similar compounds that undergo common metabolic reactions using both conventional flow electrospray ionization LC-MS and low-flow captive spray ionization (CSI) LC-MS. The differences in standard curves for four different types of transformations (O-demethylation, N-demethylation, aromatic hydroxylation, and benzylic hydroxylation) are presented. The results demonstrate that the signals of the substrates compared with those of the metabolites are statistically different in 18 of the 20 substrate-metabolite combinations for both methods. The ratio of the slopes of the standard curves varied up to 4-fold but was slightly less for the CSI method. |
| [67] |
Rappel C, Schaumloffel D. J. Anal. At. Spectrom., 2010,25(12):1963. http://xlink.rsc.org/?DOI=c0ja00050g
DOI: 10.1039/c0ja00050g |
| [68] |
Giusti P, Lobinski R, Szpunar J, Schaumloffel D. Anal. Chem., 2006,78(3):965. https://pubs.acs.org/doi/10.1021/ac051656j
DOI: 10.1021/ac051656j |
| [69] |
Shen L H, Sun J N, Cheng H Y, Liu J H, Xu Z G, Mu J X. J. Anal. At. Spectrom., 2015,30(9):1927. http://xlink.rsc.org/?DOI=C5JA00198F
DOI: 10.1039/C5JA00198F |
| [70] |
Zhou Y M, Zhang N, Li Y F, Xiong C Q, Chen S M, Chen Y T, Nie Z X. Analyst, 2014,139(21):5387. http://dx.doi.org/10.1039/c4an00979g
DOI: 10.1039/c4an00979g Better sensitivity and interface of ambient sampling/ionization mass spectrometry remain a challenge. Herein, a novel, plasma-based, ambient sampling/ionization/transmission (PASIT) integrated source in a pin-to-funnel configuration was developed for the sensitive analysis of complex samples. With the funnel sleeve directly affected by direct-current discharge plasma, PASIT combines the ability to sample/ionize analyte molecules and then efficiently collect/transport charged mass species under atmospheric pressure and consequently shows an improved sensitivity. The integrated source enhances the signal intensity by more than 2 orders of magnitude compared with the previous pin-to-plate plasma source without significant background addition. A surface limit of detection (LOD) of 130 fmol mm (2) (S/N = 3) has been achieved for clenbuterol on filter paper with an argon carrier gas. Demonstrated applications include the direct determination of active ingredients from drugs and symbolic compounds from natural plants and cholesterol from mouse brain tissue sections. |
| [71] |
Ai W P, Nie H G, Song S Y, Liu X Y, Bai Y, Liu H W. J. Am. Soc. Mass Spectrom., 2018,29(7):1408. https://doi.org/10.1007/s13361-018-1949-3
|
/
| 〈 |
|
〉 |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}