
文章编号: 20190403
文献标识码: A
局域表面等离子体共振效应在光催化技术中的应用
收稿日期:2018-08-15
要求修回日期:2018-11-22
网络出版日期:2019-01-14
基金资助
国家自然科学基金项目(21473082)
云南省科技厅第18批中青年学术带头人后备人才项目(2015HB015)
版权
Applications of Localized Surface Plasmon Resonance Effect in Photocatalysis
Received:15 Aug. 2018
rev-requestrev-request:22 Nov. 2018
Online:14 Jan. 2019
Fund
National Natural Science Foundation of China(21473082)
18th Yunnan Province Young Academic and Technical Leaders Reserve Talent Project(2015HB015)
Copyright
表面等离子激元是物理效应在光催化技术应用中的典型代表之一,作为新型光场调控技术为光催化技术的发展开辟了新的方向和思路,能够从全新的角度解决光催化技术的发展瓶颈,在过去十年来得到了广泛的研究。局域表面等离子体共振效应能够通过调节纳米颗粒的组成、形貌和介质环境等因素调控光催化体系的光谱响应范围。除此之外还能够通过增强光散射、热电子注入、诱导产生强烈的局域电场、加热周围环境等方法来增加光催化剂的氧化-还原反应速度、物质传输以及极化光催化材料表面的吸附分子,从而进一步增强材料的光催化性能。将这些优势集成到光催化材料体系中,能够显著提高传统光催化材料的太阳能转换效率,这是一个非常值得关注的发展方向。本文综述了局域表面等离子体共振效应在光催化技术中应用的基本原理、调控规律和应用等方面的研究进展,着重讨论了热电子的产生和迁移过程,贵金属中带间跃迁和表面等离子体共振效应的制约关系。最后,总结了表面等离子体光催化剂所面临的问题和挑战,并进行了相应的研究展望。
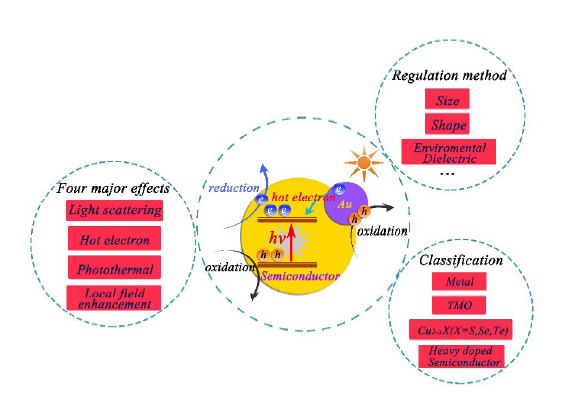
关键词: 局域表面等离子体共振 ; 光催化 ; 贵金属 ; 非贵金属 ; 缺陷半导体
中图分类号: O643;O646.9 ()
姚国英 , 刘清路 , 赵宗彦 . 局域表面等离子体共振效应在光催化技术中的应用[J]. 化学进展, 2019 , 31(4) : 516 -535 . DOI: 10.7536/PC180810
Guoying Yao , Qinglu Liu , Zongyan Zhao . Applications of Localized Surface Plasmon Resonance Effect in Photocatalysis[J]. Progress in Chemistry, 2019 , 31(4) : 516 -535 . DOI: 10.7536/PC180810
The surface plasmon effect is a typical representative of the application of physical effects in photocatalysis technology. And as new control technology of light field, it has opened up new directions and new ideas for the development of photocatalysis technology. The bottleneck of the development of photocatalytic technology can be solved from a new perspective, and has been extensively studied in the past decade. The localized surface plasmon resonance effect can regulate the spectral response range of the photocatalytic system by adjusting the composition, morphology and medium environment of the nanoparticles. In addition, the photocatalyst redox reaction rate, mass transfer, and adsorbed molecules on the surface of the polarized photocatalytic material can be increased by enhancing light scattering, hot electron injection, inducing a strong local electric field, and heating the surrounding environment, thereby further enhancing the photocatalytic properties of the material. Integrating these advantages into a photocatalytic material system can significantly improve the solar energy conversion efficiency of conventional photocatalytic materials, which is a very interesting development direction. In this review, the basic principles, material composition, regulation and recent progress of surface plasmon resonance in photocatalytic systems are presented in detail. Not only the process of generation and migration of hot electrons, but also the relationship between interband transition and surface plasmon resonance in noble metals is discussed. Finally, the prospective and challenges for future development of plasmonic photocatalysis are summarized.
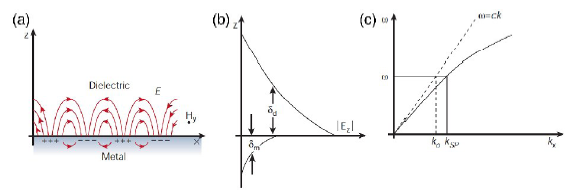
图1 表面等离子激元机理图:(a)SPP机理图;(b)SPP的电场在垂直方向呈指数递减;(c)SPP的色散曲线[40]Fig. 1 The principle of surface plasmons polaritions.(a) SPP mechanism diagram;(b) the electric field of SPP decreases exponentially in the vertical direction;(c) dispersion curve of Surface Plasmon Polariton [40]. Copyright 2003, Springer Nature. |
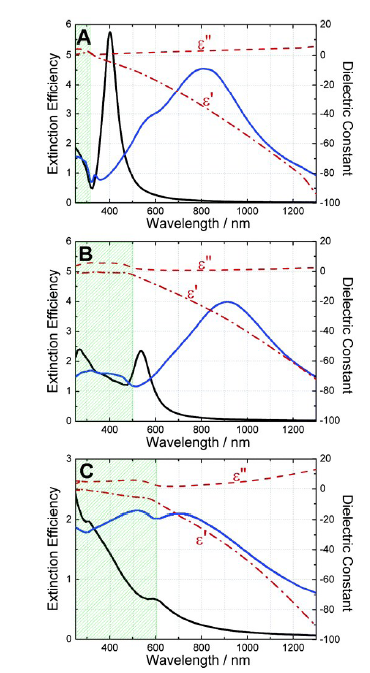
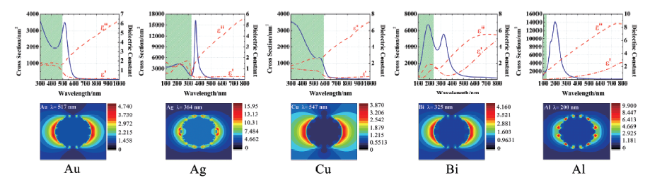
图6 理论计算的纳米球(黑实线)和纳米壳(蓝实线)的远场消光效率:(A)Ag;(B)Au;(C)Cu;ε'和ε″分别为材料介电函数的实部与虚部[61]Fig. 6 Theoretically calculate the far-field extinction efficiency of nanospheres(black solid lines) and nanoshells(blue solid lines):(A) Ag;(B) Au;(C) Cu; ε'and ε″ are the real and imaginary parts of material dielectric functions, respectively [61]. Copyright 2005, American Chemical Society. |
图8 在非相互作用的情况下纳米球电场强度的空间分布:(a)Drude模型;(b)洛伦兹模型;(c~e)Drude-洛伦兹模型在强相互作用情况下;(f)Drude(红线),洛伦兹(绿线)和DL模型(黑线)描述的纳米球的光吸收谱[62]Fig. 8 Spatial distributions of the E-field intensity(|E|2) of a nanosphere(a=10 nm) described by(a) the Drude model and(b) the Lorentz model in the noninteracting cases as well as(c~e) the DL model in the strong interaction case.(f) Optical absorption spectra of the nanospheres described by the Drude(red line), Lorentz(green line), and DL models(dark line)[62]. Copyright 2011, American Chemical Society. |
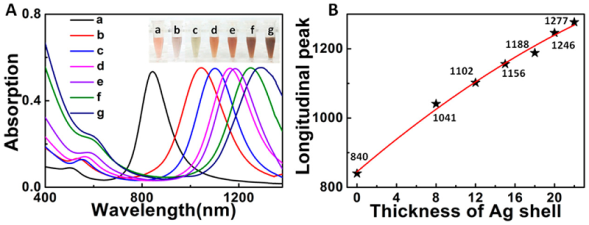
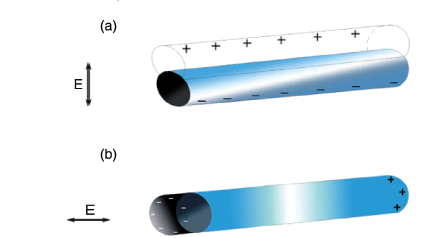
图10 具有不同不连续Ag壳厚度的双金属Au/Ag核-壳超结构的(A)代表性UV-vis-NIR吸收光谱:(a)8,(b)12,(c)15,(d)18,(e) 20,和(f)22 nm;插图为相应的溶液颜色照片。(B)纵向吸收峰强度与Ag壳厚度的关系[82]Fig. 10 Representative UV-vis-NIR absorption spectra of bimetallic Au/Ag core-shell superstructures;(A) with different discontinuous Ag shell thicknesses:(a) 8,(b) 12,(c) 15,(d) 18,(e) 20, and(f) 22 nm; corresponding inset photographs of solution color.(B) Plot of longitudinal absorption peak intensity versus thickness of Ag shell[82]. Copyright 2017, American Chemical Society. |
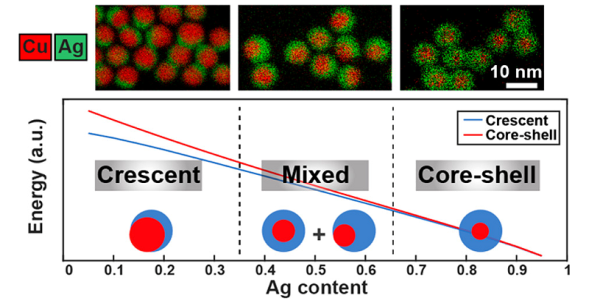
图11 以新月结构是平衡结构的条件下,对于Ag不同原子分数计算三种可能的Cu-Ag形状的能量,这种划分与实验结果相匹配[86]Fig. 11 Calculated energies of three possible Cu-Ag shapes for different atomic fractions of Ag under the condition where the crescent is the equilibrium structure, such division matches the experimentally obtained results [86]. Copyright 2018, American Chemical Society. |
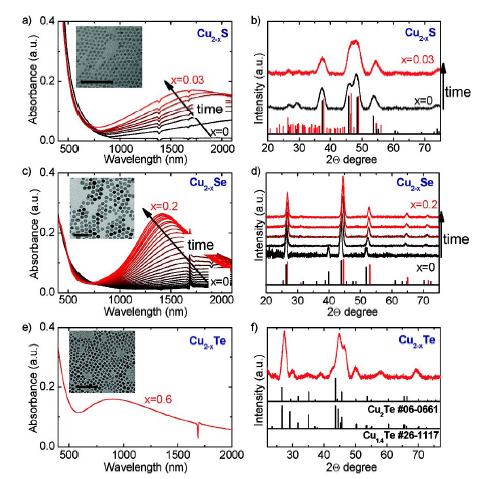
图14 空气暴露氧化过程中化学计量在甲苯中的可见-近红外消光谱的时间演变图和XRD图谱的演变。 (a、c、e)分别为Cu2-xS(x=0,黑色曲线)、Cu2-xSe(x=0,黑色曲线)和Cu2-xTe(x> 0)纳米晶体在甲苯中的Vis-NIR消光光谱。(b、d、f)为氧化期间(a、c、e)中所示的Cu2-xS、Cu 2-xSe、Cu2-xTe的XRD图谱的时间演变[116]Fig. 14 The time evolution of the visible-near-infrared spectroscopy of stoichiometry in toluene during air exposure oxidation and the evolution of the XRD patterns. Nanocrystals in Vis-NIR in toluene Extinction spectrum(a) Cu2-xS(x=0, black curve),(c) Cu2-xSe(x=0, black curve) and(e) Cu2-xTe(x > 0). The time evolution of the XRD patterns of(b) Cu2-xS,(d) Cu2-xSe,(f) Cu2-xTe shown in the oxidation period(a, c, e) [116]. Copyright 2012, American Chemical Society. |
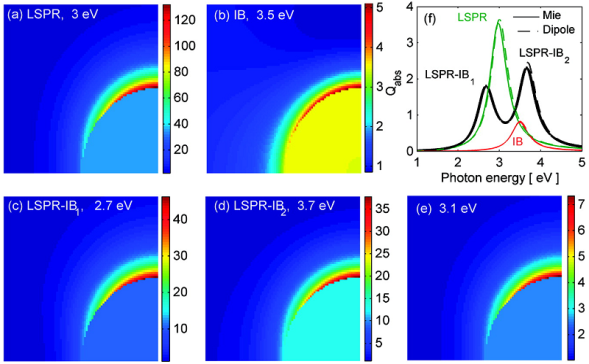
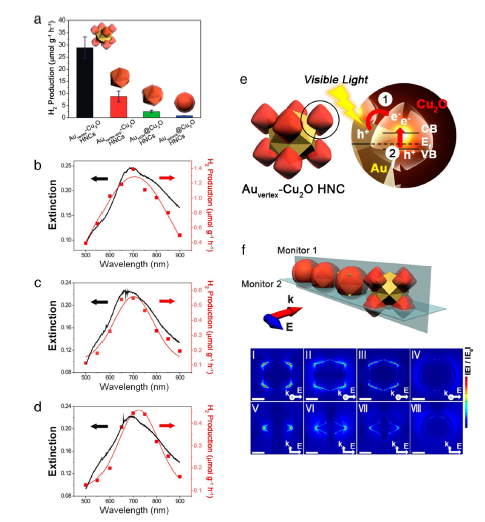
图19 (a)不同异质纳米晶体的光催化氢生成速率。用(b)Auvertex-Cu2O,(c)Auvertex-exp-Cu2O和(d)AuHOH @Cu2O 异质纳米晶体获得的消光光谱;(e)Auvertex-Cu2O 异质纳米晶体可能的等离子体激元诱导的电荷分离过程的示意图;(f)用于计算| E |的FDTD仿真模型异质纳米晶体:Auvertex-Cu2O(Ⅰ,Ⅴ),Auvertex-exp-Cu2O(Ⅱ,Ⅵ),AuHOH @Cu2O(Ⅲ,Ⅶ)和Ausphere @Cu2O异质纳米晶体(Ⅳ,Ⅷ)[148]Fig. 19 (a) Photocatalytic hydrogen generation rate of different heterogeneous nanocrystals; The extinction spectrum obtained with(b) Auvertex-Cu2O,(c) Auvertex-exp-Cu2O and(d) AuHOH@Cu2O heterogeneous nanocrystals.(e) Schematic diagram of possible plasmon-induced charge separation processes for Auvertex-Cu2O heterogeneous nanocrystals.(f) FDTD simulation model heterogeneous nanocrystals used to calculate | E | Auvertex-Cu2O(Ⅰ,Ⅴ), Auvertex-exp-Cu2O(Ⅱ,Ⅵ), AuHOH @Cu2O(Ⅲ,Ⅶ) and Ausphere @Cu2O Heteronanocrystals(Ⅳ,Ⅷ) [148]. Copyright 2016, American Chemical Society. |
| [1] |
Fujishima A, Honda K . Nature, 1972,238(5358):37.
|
| [2] |
Ihara T, Miyoshi M, Iriyama Y, Matsumoto O, Sugihara S. Appl. Catal . B: Environ., 2003,42(4):403.
|
| [3] |
Xu M, Da P M, Wu H Y, Zhao D Y ,Zheng G F. Nano Lett, 2012,12(3):1503.
|
| [4] |
Yin G N, Ma J X, Jiang H, Li J, Yang D, Gao F, Zeng J H, Liu Z K ,Liu S D. ACS Appl Mater. Interfaces, 2017,9(12):10752.
|
| [5] |
Long R, English N J . J. Phys. Chem. C, 2009,113(21):9423.
|
| [6] |
Long R, English N J . J. Phys. Chem. C, 2010,114(27):11984.
|
| [7] |
Mowbray D J, Martinez J I, Lastra J M G, Thygesen K S, Jacobsen K W. J. . Phys. Chem. C, 2010,113(28):12301.
|
| [8] |
Song J N, Zheng M J, Yuan X L, Li Q, Wang F, Ma L G, You Y X, Liu S H, Liu P J, Jiang D K, Ma L, Shen W Z . Mater. Sci., 2017,52(12):6976.
|
| [9] |
Han X X, Huang J, Jing X X, Yang D Y, Lin H, Wang Z G, Li P, Chen Y . ACS Nano, 2018,12(5):4545.
|
| [10] |
Nishioka S, Hyodo J, Vequizo J J M, Yamashita S, Kumagai H, Kimoto K, Yamakata A, Yamazaki Y, Maeda K . ACS Catalysis, 2018,8(8):7190.
|
| [11] |
Bessekhouad Y, Robert D, Weber J V . J. Photochem. Photobiol. A: Chem., 2004,163(3):569.
|
| [12] |
Wang Y J, Wang Q S, Zhan X Y, Wang F M, Safdar M, He J . Nanoscale, 2013,5(18):8326.
|
| [13] |
Guo L, Yang Z, Marcus K, Li Z, Luo B, Zhou L, Wang X, Du Y, Yang Y. Energy Environ . Sci., 2018,11(1):106.
|
| [14] |
Gong Y J, Lin J H, Wang X L, Shi G, Lei S D, Lin Z, Zou X L, Ye G L, Vajtai R ,Yakobson B I. Nat. Mater, 2014,13(12):1135.
|
| [15] |
Zhang J, Wang J H, Chen P, Sun Y, Wu S, Jia Z Y, Lu X B, Yu H, Chen W ,Zhu J Q. Adv. Mater, 2015,28:1950
|
| [16] |
Chen H L, Wen X W, Zhang J, Wu T M, Gong Y J, Zhang X, Yuan J T, Yi C Y, Lou J ,Ajayan P M. Nat. Commun, 2016,7:12512.
|
| [17] |
Tada H, Suzuki F, Ito S, Akita T, Tanaka K, Kawahara T, Kobayashi H . J. Phys. Chem. B, 2002,106(34):8714.
|
| [18] |
Yan H J, Yang J H, Ma G J, Wu G P, Zong X, Lei Z B, Shi J Y, Li C . Catal., 2009,266(2):165.
|
| [19] |
Foo W J, Zhang C, Ho G W . Nanoscale, 2013,5(2):759.
|
| [20] |
An B, Zhang J Z, Cheng K, Ji P F, Wang C, Lin W B . Am. Chem. Soc., 2017,139(10):3834. https://www.ncbi.nlm.nih.gov/pubmed/28209054
DOI: 10.1021/jacs.7b00058 PMID: 28209054 2 play vital roles in the hydrogenation of CO2 to methanol by these composite catalysts. Surface structural reorganization and particle growth during catalysis deleteriously reduce these active interfaces, diminishing both catalytic activities and MeOH selectivities. Here we report the use of preassembled bpy and Zr6(μ3-O)4(μ3-OH)4 sites in UiO-bpy metal-organic frameworks (MOFs) to anchor ultrasmall Cu/ZnOx nanoparticles, thus preventing the agglomeration of Cu NPs and phase separation between Cu and ZnOx in MOF-cavity-confined Cu/ZnOx nanoparticles. The resultant Cu/ZnOx@MOF catalysts show very high activity with a space-time yield of up to 2.59 gMeOH kgCu-1 h-1, 100% selectivity for CO2 hydrogenation to methanol, and high stability over 100 h. These new types of strong metal-support interactions between metallic nanoparticles and organic chelates/metal-oxo clusters offer new opportunities in fine-tuning catalytic activities and selectivities of metal nanoparticles@MOFs.]]> |
| [21] |
Chau L K, Lin Y F, Cheng S F ,Lin T J. Sens. Actuator. B: Chem., 2006,113(1):100.
|
| [22] |
Homola J . Chem.Rev., 2008,108(2):462.
|
| [23] |
Zhang S H, Huang Q, Zhang L J, Zhang H, Han Y B, Sun Q, Cheng Z X, Qin H Z, Dou S X, Li Z . Nanoscale, 2018,10(7):3130.
|
| [24] |
Wang F F, Wang H J, Liu X Y, Wu D P, Jiang K, Li Q ,Xu D S. Adv. Energy Mater, 2018,8(20):1800136.
|
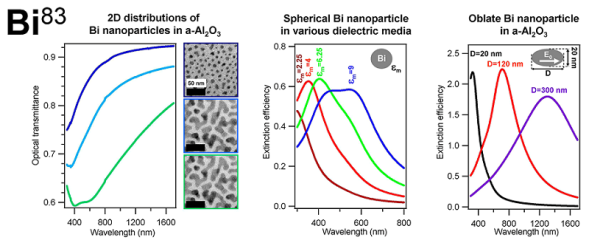
| [25] |
Kardarian K, Nunes D, Maria Sberna P, Ginsburg A, Keller D A, Vaz Pinto J, Deuermeier J, Anderson A Y, Zaban A, Martins R ,Fortunato E. Sol. Energy Mater. Sol. Cells, 2016,147:27.
|
| [26] |
Awazu K, Fujimaki M, Rockstuhl C, Tominaga J, Murakami H, Ohki Y, Yoshida N, Watanabe T . Am. Chem. Soc., 2008,130(5):1676. https://www.ncbi.nlm.nih.gov/pubmed/18189392
DOI: 10.1021/ja076503n PMID: 18189392 Titanium dioxide (TiO2) displays photocatalytic behavior under near-ultraviolet (UV) illumination. In another scientific field, it is well understood that the excitation of localized plasmon polaritons on the surface of silver (Ag) nanoparticles (NPs) causes a tremendous increase of the near-field amplitude at well-defined wavelengths in the near UV. The exact resonance wavelength depends on the shape and the dielectric environment of the NPs. We expected that the photocatalytic behavior of TiO2 would be greatly boosted if it gets assisted by the enhanced near-field amplitudes of localized surface plasmon (LSP). Here we show that this is true indeed. We named this new phenomenon "plasmonic photocatalysis". The key to enable plasmonic photocatalysis is to deposit TiO2 on a NP comprising an Ag core covered with a silica (SiO2) shell to prevent oxidation of Ag by direct contact with TiO2. The most appropriate diameter for Ag NPs and thickness for the SiO2 shell giving rise to LSP in the near UV were estimated from Mie scattering theory. Upon implementing a device that took these design considerations into account, the measured photocatalytic activity under near UV illumination of such a plasmonic photocatalyst, monitored by decomposition of methylene blue, was enhanced by a factor of 7. The enhancement of the photocatalytic activity increases with a decreased thickness of the SiO2 shell. The plasmonic photocatalysis will be of use as a high performance photocatalyst in nearly all current applications but will be of particular importance for applications in locations of minimal light exposure. |
| [27] |
Wang P, Huang B B, Qin X Y, Zhang X Y, Dai Y, Wei J Y ,Whangbo M H. Angew. Chem. Int. Edi., 2008,47(41):7931.
|
| [28] |
Wang P, Huang B B, Zhang X Y, Qin X Y, Jin H, Dai Y, Wang Z Y, Wei J Y, Zhan J, Wang S Y, Wang J P, Whangbo M H . Chem. -A Eur. J., 2009,15(8):1821.
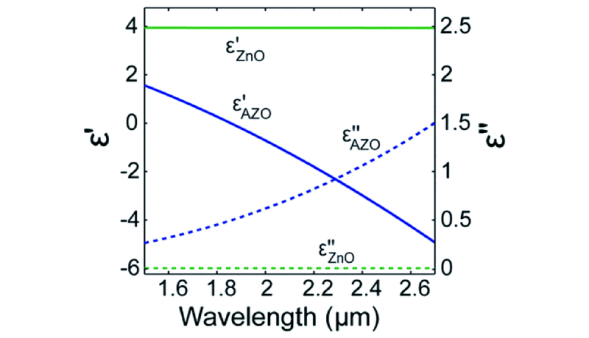
|
| [29] |
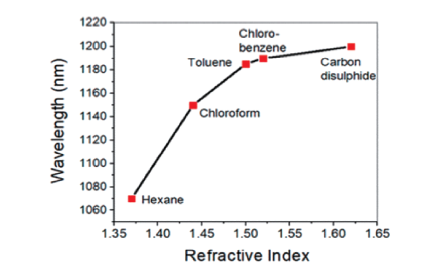
Chen X, Zhu H Y, Zhao J C, Zheng Z F ,Gao X P. Angew. Chem. Int Edit., 2008,47(29):5353.
|
| [30] |
Jain P K, Lee K S , El-Sayed I H, El-Sayed M A . J. Phys. Chem. B, 2006,110(14):7238.
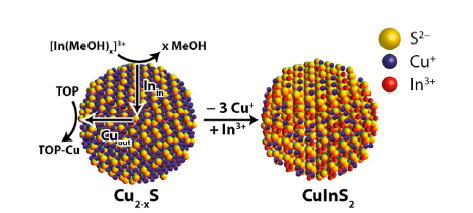
|
| [31] |
Linic S, Christopher P ,Ingram D B. Nat. Mater, 2011,10(12):911.
|
| [32] |
Zhou C G, Wang S M, Zhao Z Y, Shi Z, Yan S C ,Zou Z G. Adv. Funct. Mater, 2018,28(31):1801214.
|
| [33] |
Yang J H, Guo Y Z, Jiang R B, Qin F, Zhang H, Lu W Z, Wang J F, Yu J C . Am. Chem. Soc., 2018,140(27):8497. https://www.ncbi.nlm.nih.gov/pubmed/29905477
DOI: 10.1021/jacs.8b03537 PMID: 29905477 2 to NH3 is an essential process for sustaining life. One grand challenge is to develop efficient catalysts to photofix N2 under ambient conditions. Herein we report an all-inorganic catalyst, Au nanocrystals anchored on ultrathin TiO2 nanosheets with oxygen vacancies. It can accomplish photodriven N2 fixation in the "working-in-tandem" pathway at room temperature and atmospheric pressure. The oxygen vacancies on the TiO2 nanosheets chemisorb and activate N2 molecules, which are subsequently reduced to NH3 by hot electrons generated from plasmon excitation of the Au nanocrystals. The apparent quantum efficiency of 0.82% at 550 nm for the conversion of incident photons to NH3 is higher than those reported so far. Optimizing the absorption across the overall visible range with the mixture of Au nanospheres and nanorods further enhances the N2 photofixation rate by 66.2% in comparison with Au nanospheres used alone. This work offers a new approach for the rational design of efficient catalysts toward sustainable N2 fixation through a less energy-demanding photochemical process compared to the industrial Haber-Bosch process.]]> |
| [34] |
Patnaik S, Swain G, Parida K M . Nanoscale, 2018,10(13):5950.
|
| [35] |
Zeng Z P, Li T, Li Y B, Dai X C, Huang M H, He Y H, Xiao G C, Xiao F X . J. Mater. Chem. A, 2018, DOI: 10.1039/C8TA08841A.
|
| [36] |
Lou Z Z, Wang Z Y, Huang B B, Dai Y . ChemCatChem, 2014,6(9):2456.
|
| [37] |
Wang Z Y, Liu Y Y, Huang B B, Dai Y, Lou Z Z, Wang G, Zhang X Y ,Qin X Y. Phys. Chem. Chem Phys., 2014,16(7):2758.
|
| [38] |
Liu L Q, Zhang X N, Yang L F, Ren L T, Wang D F ,Ye J H. Nat. Sci. Rev, 2017,4(5):761.
|
| [39] |
Zhang N, Han C, Fu X Z, Xu Y J . Chem, 2018,4(8):1832.
|
| [40] |
Barnes W L, Dereux A, Ebbesen T W . Nature, 2003,424:824.
|
| [41] |
Ding S Y, Yi J, Li J F, Ren B, Wu D Y, Panneerselvam R ,Tian Z Q. Nat. Rev. Mater, 2016,1:16021.
|
| [42] |
Kelly K L, Coronado E, Zhao L L, Schatz G C . J. Phys. Chem. B, 2003,107(3):668.
|
| [43] |
Sambles J R, Bradbery G W, Yang F . Contemp. Phys 1991,32(3):173.
|
| [44] |
Eustis S, ,El-Sayed M A. Chem. Soc. Rev., 2006,35(3):209.
|
| [45] |
Barolo G, Livraghi S, Chiesa M, Paganini M C, Giamello E . J. Phys. Chem. C, 2012,116(39):20887.
|
| [46] |
Linic S, Aslam U, Boerigter C, Morabito M . Nat. Mater 2015,14(6):567.
|
| [47] |
Low J X, Qiu S Q, Xu D F, Jiang C J ,Cheng B. Appl. Surf. Sci, 2018,434:423.
|
| [48] |
Li M, Xing Z P, Jiang J J, Li Z Z, Yin J W, Kuang J Y, Tan S Y, Zhu Q, Zhou W . Taiwan Inst. Chem. Eng., 2018,82:198.
|
| [49] |
Tanaka A, Hashimoto K, Kominami H . Chem. Commun 2017,53(35):4759.
|
| [50] |
Jiao Z B, Shang M D, Liu J M, Lu G X, Wang X S, Bi Y P . Nano Energy, 2017,31:96.
|
| [51] |
Cushing S K, Li J, Meng F, Senty T R, Suri S, Zhi M, Li M, Bristow A D, Wu N . J Am Chem Soc, 2012,134(36):15033.
|
| [52] |
Lu B, Liu A P, Wu H P, Shen Q P, Zhao T Y, Wang J S . Langmuir, 2016,32(12):3085.
|
| [53] |
Li J T, Cushing S K, Bright J, Meng F, Senty T R, Zheng P, Bristow A D ,Wu N Q. ACS Catal, 2014,3(1):47.
|
| [54] |
Ma X C, Dai Y, Yu L, Huang B B . Light: Sci. Appl., 2016,5:e16017.
|
| [55] |
Christopher P, Xin H, Linic S . Nat. Chem 2011,3:467.
|
| [56] |
Liu L C, Ji Z Y, Zou W X, Gu X R, Deng Y, Gao F, Tang C J, Dong L . ACSCatal., 2013,3(9):2052.
|
| [57] |
Meng X G, Wang T, Liu L Q, Ouyang S X, Li P, Hu H L, Kako T, Iwai H, Tanaka A ,Ye J H. Angew. Chem. Int. Edit., 2014,53(43):11478.
|
| [58] |
Liu H M, Meng X G, Dao T D, Zhang H B, Li P, Chang K, Wang T, Li M, Nagao T ,Ye J H. Angew. Chem. Int. Edit., 2015,54(39):11545.
|
| [59] |
Schelm S ,Smith G B. Appl. Phys. Lett, 2003,82(24):4346.
|
| [60] |
Chen C J ,Chen D H. Chem. Eng. J, 2012,180(6):337.
|
| [61] |
Wang H, Tam F, Grady N K, Halas N J . J. Phys. Chem. B, 2005,109(39):18218.
|
| [62] |
Pakizeh T . J. Phys. Chem. C, 2011,115(44):21826.
|
| [63] |
Zhang Z S, Liu L H, Fang W H, Long R, Tokina M V, Prezhdo O V . Chem, 2018,4(5):1112.
|
| [64] |
Long R, Prezhdo O V . Am. Chem. Soc., 2014,136(11):4343. https://www.ncbi.nlm.nih.gov/pubmed/24568726
DOI: 10.1021/ja5001592 PMID: 24568726 Photoexcitation of the plasmon band in metallic nanoparticles adsorbed on a TiO2 surface initiates many important photovoltaic and photocatalytic processes. The traditional view on the photoinduced charge separation involves excitation of a surface plasmon, its subsequent dephasing into electron-hole pairs, followed by electron transfer (ET) from the metal nanoparticle into TiO2. We use nonadiabatic molecular dynamics combined with time-domain density functional theory to demonstrate that an electron appears inside TiO2 immediately upon photoexcitation with a high probability (~50%), bypassing the intermediate step of electron-hole thermalization inside the nanoparticle. By providing a detailed, atomistic description of the charge separation, energy relaxation, and electron-hole recombination processes, the simulation rationalizes why the experimentally observed ultrafast photoinduced ET in an Au-TiO2 system is possible in spite of the fast energy relaxation. The simulation shows that the photogenerated plasmon is highly delocalized onto TiO2, and thus, it is shared by the electron donor and acceptor materials. In the 50% of the cases remaining after the instantaneous photogeneration of the charge-separated state, the electron injects into TiO2 on a sub-100 fs time scale by the nonadiabatic mechanism due to high density of acceptor states. The electron-phonon relaxation parallels the injection and is slower, resulting in a transient heating of the TiO2 surface by 40 K. Driven by entropy, the electron moves further into TiO2 bulk. If the electron remains trapped at the TiO2 surface, it recombines with the hole on a picosecond time scale. The obtained ET and recombination times are in excellent agreement with the experiment. The delocalized plasmon state observed in our study establishes a novel concept for plasmonic photosensitization of wide band gap semiconductors, leading to efficient conversion of photons to charge carriers and to hybrid materials with a wide variety of applications in photocatalysis and photovoltaics. |
| [65] |
Powell C J . Phys. Rev., 1965,15(22):852.
|
| [66] |
Powell C J . Phys. Rev., 1968,175(3):972.
|
| [67] |
Hagemann H J, Gudat W, Kunz C . Opt. Soc. Am., 1975,65(6):742.
|
| [68] |
Sugawa K, Yamaguchi D, Tsunenari N, Uchida K, Tahara H, Takeda H, Tokuda K, Jin S, Kusaka Y, Fukuda N. ACS Appl . Mater. Interfaces, 2016,9(1):750.
|
| [69] |
Sugawa K, Tsunenari N, Takeda H, Fujiwara S, Akiyama T, Honda J, Igari S, Inoue W, Tokuda K, Takeshima N, Watanuki Y, Tsukahara S, Takase K, Umegaki T, Kojima Y, Nishimiya N, Fukuda N, Kusaka Y, Ushijima H, Otsuki J . Langmuir, 2017,33(23):5685.
|
| [70] |
Mcmahon J M, Schatz G C ,Gray S K. Phys. Chem. Chem. Phys., 2013,15(15):5415.
|
| [71] |
Xiao F X, Zeng Z P, Liu B . Am. Chem. Soc., 2015,137(33):10735. https://www.ncbi.nlm.nih.gov/pubmed/26258281
DOI: 10.1021/jacs.5b06323 PMID: 26258281 In recent years, enormous attention has been paid to the construction of metal cluster-semiconductor nanocomposites because of the fascinating and unique properties of metal clusters; however, investigations on photoelectrochemical (PEC) and photocatalytic properties of metal cluster-semiconductor systems are still rare. Moreover, to date, intrinsic correlation between metal clusters and bulk metal nanocrystals has yet to be elucidated. In this work, a facile layer-by-layer (LbL) self-assembly strategy has been developed to judiciously and intimately integrate gold nanocrystals (Au) within the interface between gold clusters (Au(x)) and hierarchically ordered TiO2 nanotube arrays framework, by which imperative roles of Au nanocrystals as electron relay mediator and plasmonic sensitizer for Aux clusters were revealed. In addition, it was found that synergistic interaction between Au nanocrystals and Aux clusters contributed to promising visible-light-driven photocatalytical and PEC performances. It is anticipated that our work could provide a general way for rationally constructing metal and metal clusters codecorated semiconductor heterostructures and, more significantly, bridge the gap between metal clusters and metal nanocrystals for a diverse range of applications. |
| [72] |
Gao Y, Lin J Y, Zhang Q Z, Yu H, Ding F, Xu B T, Sun Y G ,Xu Z H. Appl. Catal. B: Environ., 2018,224:586.
|
| [73] |
Cheng W R, Su H, Tang F M, Che W, Huang Y Y, Zheng X S, Yao T, Liu J K, Hu F C, Jiang Y, Liu Q H, Wei S Q . J. Mater. Chem. A, 2017,5(37):19649.
|
| [74] |
Kumari G, Zhang X Q, Devasia D, Heo J, Jain P K . ACS Nano, 2018,12(8):8330.
|
| [75] |
Liu L Q, Ouyang S X ,Ye J H. Angew. Chem, 2013,125(26):6821.
|
| [76] |
Liu L Q, Dao T D, Kodiyath R, Kang Q, Abe H, Nagao T ,Ye J H. Adv. Funct. Mater, 2014,24(48):7754.
|
| [77] |
Zhu M S, Cai X Y, Fujitsuka M, Zhang J Y ,Majima T. Angew. Chem. Int Edit., 2017,129(8):2096.
|
| [78] |
Zhang Z Y, Cao S W, Liao Y S, Xue C. Appl. Catal . B: Environ., 2015,162:204.
|
| [79] |
Verma P, Yuan K, Kuwahara Y, Mori K, Yamashita H. Appl. Catal . B: Environ., 2018,223:10.
|
| [80] |
Zhou Y X, Wang D S ,Li Y D. Chem. Commun, 2014,50(46):6141.
|
| [81] |
Cui Q L, Shen G Z, Yan X H, Li L D, Möhwald H, Bargheer M. ACS Appl . Mater. Interfaces, 2014,6(19):17075.
|
| [82] |
Dai L W, Song L P, Huang Y J, Zhang L, Lu X F, Zhang J W, Chen T . Langmuir, 2017,33(22):5378.
|
| [83] |
Pellarin M, Issa I, Langlois C, Lebeault M A, Ramade J, Lermé J, Broyer M, Cottancin E J . J. Phys. Chem. C, 2015,119(9):5002.
|
| [84] |
Govorov A O, Zhang H, Gun’ko Y K . J. Phys. Chem. C, 2013,117(32):16616.
|
| [85] |
Liu L Q, Li P, Adisak B, Ouyang S, Umezawa N, Ye J H, Kodiyath R, Tanabe T, Ramesh G V, Ueda S . J. Mater. Chem. A, 2014,2(25):9875.
|
| [86] |
Osowiecki W T, Ye X C, Satish P, Bustillo K C, Clark E L, Alivisatos A P . Am. Chem. Soc., 2018,140(27):8569. https://www.ncbi.nlm.nih.gov/pubmed/29909616
DOI: 10.1021/jacs.8b04558 PMID: 29909616 2O upon mild oxidation, whereas fully core-shell Cu@Ag NCs are robust against oxidation up to 100 °C. The plasmonic and interband absorptions of Cu-Ag NCs depend on the composition and the degree of Cu oxidation, which may find application in light-driven catalysis.]]> |
| [87] |
Wang P, Huang B B, Lou Z Z, Zhang X Y, Qin X Y, Dai Y, Zheng Z K, Wang X N . Chem.-A Eur. J., 2009,16(2):538.
|
| [88] |
Cheng H F, Huang B B, Dai Y, Qin X Y, Zhang X Y . Langmuir, 2010,26(9):6618.
|
| [89] |
Liang X Z, Wang P, Li M M, Zhang Q Q, Wang Z Y, Dai Y, Zhang X Y, Liu Y Y, Whangbo M H ,Huang B B. Appl. Catal. B: Environ., 2018,220:356.
|
| [90] |
Ye L Q, Liu J Y, Gong C Q, Tian L H, Peng T Y, Zan L. ACS Catal ., 2012,2(8):1677.
|
| [91] |
Zhang P Y, Song T, Wang T T ,Zeng H P. RSC Adv, 2017,7(29):17873.
|
| [92] |
Zhang P Y, Song T, Wang T T ,Zeng H P. Appl. Catal. B: Environ., 2018,225:172.
|
| [93] |
Zhang P Y, Song T, Wang T T ,Zeng H P. Int. J. Hydrogen Energy, 2017,42(21):14511.
|
| [94] |
Zhang P Y, Wang T T ,Zeng H P. Appl. Surf. Sci, 2017,391:404.
|
| [95] |
Gavade N L, Babar S B, Kadam A N, Gophane A D ,Garadkar K M. Ind. Eng. Chem. Res., 2017,56(49):14489.
|
| [96] |
Yang L, Pillai S ,Green M A. Sci. Rep, 2015,5:11852.
|
| [97] |
He W J, Sun Y J, Jiang G M, Huang H W, Zhang X M, Dong F. Appl. Catal . B: Environ., 2018,232:340.
|
| [98] |
He W J, Sun Y J, Jiang G M, Li Y H, Zhang X M, Zhang Y X, Zhou Y, Dong F. Appl. Catal . B: Environ., 2018,239; 619.
|
| [99] |
Lv Y H, Cao X F, Jiang H Y, Song W J, Chen C C ,Zhao J C. Appl. Catal.B:Environ., 2016,194:150.
|
| [100] |
Cheng Y H, Lin Y J, Xu J P, He J, Wang T Z, Yu G J, Shao D W, Wang W H, Lu F, Li L, Du X, Wang W C, Liu H ,Zheng R K. Appl. Surf. Sci, 2016,366:120.
|
| [101] |
Nie J ,Patrocinio A O T, Hamid S, Sieland F, Sann J, Xia S, Bahnemann D W , Schneider. Phys. Chem. Chem. Phys., 2018,20(7):5264. https://www.ncbi.nlm.nih.gov/pubmed/29400385
DOI: 10.1039/c7cp07762a PMID: 29400385 2 nanoparticles (Cu-TiO2) using different methods aiming at the production of highly efficient visible light photocatalysts. Photocatalytic H2 evolution rates obtained from methanol/water mixtures revealed no significant influence of the presence of copper oxides on the photoreaction upon visible light illumination. The photocatalytic H2 production rates were evaluated upon illumination with different spectral ranges (≥420 nm or ≥500 nm) and the results evidenced that the visible light induced charge carrier formation on the Cu-TiO2 photocatalysts consists of two distinct pathways: the direct excitation of TiO2 and the induced excitation by the so-called surface plasmon resonance (SPR) effect of the Cu nanoparticles on the TiO2 surface. Both pathways are present when the full visible range of the spectrum is used (≥420 nm), while for illumination at longer wavelengths (≥500 nm), the photocatalytic activity is solely promoted by the Cu-SPR effect. Electron paramagnetic resonance (EPR) and laser flash photolysis measurements were performed to clarify the underlying mechanism of Cu-TiO2 photocatalysts upon visible light illumination.]]> |
| [102] |
Toudert J, Serna R ,Jiménez De Castro M. J. Phys. Chem. C, 2012,116(38):20530.
|
| [103] |
Sun Y J, Zhao Z W, Zhang W D, Gao C F, Zhang Y X, Dong F . Colloid Interface Sci., 2017,485:1. https://www.ncbi.nlm.nih.gov/pubmed/27639168
DOI: 10.1016/j.jcis.2016.09.018 PMID: 27639168 2CO3 (Bi-BOC) nanocomposites and Bi elemental photocatalysts. The Bi nanoparticles were produced via the insitu reduction of (BiO)2CO3 by NaBH4. The catalysts were utilized for the photocatalytic NO removal under visible light and UV illumination. Significantly, the photocatalytic capability of the Bi-BOC was highly enhanced with an unprecedented NO removal of 63.6%. The Bi metal demonstrated a direct plasmonic photocatalytic NO removal ratio of 53.6% under UV irradiation. The significantly enhanced photocatalytic capability of Bi-BOC can be ascribed to the synergistic effects of the SPR effect, enhanced visible-light-harvesting and the efficient electron-hole separation induced by Bi nanoparticles. The Bi nanoparticles can perform as a non-noble metal-based plasmonic cocatalyst for advancing photocatalytic ability. The mechanism of photocatalytic NO oxidation was proposed and compared under both visible light and UV illumination. Furthermore, the Bi-BOC photocatalysts showed good photochemical stability under repeated tests. This work could not only offer new insights into in-situ fine-tune reduction strategy for Bi-based photocatalysts, but also proves the potentials of utilizing low cost Bi cocatalysts as a substitute for noble metals to improve other photocatalysts.]]> |
| [104] |
Li X W, Zhang W D, Cui W, Sun Y J, Jiang G M, Zhang Y X, Huang H W, Dong F. Appl. Catal . B: Environ., 2018,221:482.
|
| [105] |
Wang H, Zhang W D, Li X W, Li J Y, Cen W L, Li Q Y, Dong F. Appl. Catal . B: Environ., 2018,225:218.
|
| [106] |
Yang F, Zhu X M, Fang J Z, Chen D D, Feng W H ,Fang Z Q. Ceram. Int, 2018,44(6):6918.
|
| [107] |
Chen D D, Wu S X, Fang J Z, Lu S Y, Zhou G Y, Feng W H, Yang F, Chen Y ,Fang Z Q. Sep. Purif. Technol, 2018,193:232.
|
| [108] |
Hu J L, Chen L, Lian Z C, Cao M, Li H J, Sun W B, Tong N L, Zeng H B . J. Phys. Chem. C, 2012,116(29):15584.
|
| [109] |
Ahmadivand A ,Golmohammadi S. Opt. Laser Technol, 2015,66:9.
|
| [110] |
Ghori M Z, Veziroglu S, Hinz A, Shurtleff B B, Polonskyi O, Strunskus T, Adam J, Faupel F ,Aktas O C. ACS Appl. Nano Mater, 2018, DOI: 10.1021/acsanm.8b00853.
|
| [111] |
Liu Y, Liu M X, Swihart M T . J. Phys. Chem. C, 2017,121(25):13435.
|
| [112] |
Lin R, Wan J W, Xiong Y, Wu K L, Cheong W C, Zhou G, Wang D S, Peng Q, Chen C, Li Y D . Am. Chem. Soc., 2018,140(29):9078. https://www.ncbi.nlm.nih.gov/pubmed/29979871
DOI: 10.1021/jacs.8b05293 PMID: 29979871 3 nanosheets and nanowires with dominant exposed facets of {001} and {110}, respectively. The lower hole effective mass on {110} (0.94 m0) than on {001} (1.28 m0) calculated by density functional theory leads to the higher hole mobility on {110} (4.92 cm2 V-1 s-1) than on {001} (3.14 cm2 V-1 s-1). Combined with the Einstein equation and the lifetime of the hole, the calculated hole diffusion length on {110} (74.8 nm) is larger than on {001} (53.4 nm). Overall, the lower hole effective mass, higher hole mobility, and greater hole diffusion length on {110} collectively result in a photocatalytic activity on benzyl alcohol oxidation 2.46 times as high as that on {001}.]]> |
| [113] |
Song G S, Shen J, Jiang F R, Hu R G, Li W Y, An L, Zou R J, Chen Z G, Qin Z Y ,Hu J Q. ACS Appl Mater Interfaces, 2014,6(6):3915.
|
| [114] |
Gordon T R, Paik T, Klein D R, Naik G V, Caglayan H, Boltasseva A ,Murray C B. Nano Lett, 2013,13(6):2857.
|
| [115] |
Naik G V, Liu J J, Kildishev A V, Shalaev V M ,Boltasseva A. Proc. Natl. Acad. Sci. U. S A., 2012,109(23):8834.
|
| [116] |
Kriegel I, Jiang C, Rodríguezfernández J, Schaller R D, Talapin D V, Da C E, Feldmann J . Am. Chem. Soc., 2012,134(3):1583. https://www.ncbi.nlm.nih.gov/pubmed/22148506
DOI: 10.1021/ja207798q PMID: 22148506 The optical properties of stoichiometric copper chalcogenide nanocrystals (NCs) are characterized by strong interband transitions in the blue part of the spectral range and a weaker absorption onset up to ~1000 nm, with negligible absorption in the near-infrared (NIR). Oxygen exposure leads to a gradual transformation of stoichiometric copper chalcogenide NCs (namely, Cu(2-x)S and Cu(2-x)Se, x = 0) into their nonstoichiometric counterparts (Cu(2-x)S and Cu(2-x)Se, x > 0), entailing the appearance and evolution of an intense localized surface plasmon (LSP) band in the NIR. We also show that well-defined copper telluride NCs (Cu(2-x)Te, x > 0) display a NIR LSP, in analogy to nonstoichiometric copper sulfide and selenide NCs. The LSP band in copper chalcogenide NCs can be tuned by actively controlling their degree of copper deficiency via oxidation and reduction experiments. We show that this controlled LSP tuning affects the excitonic transitions in the NCs, resulting in photoluminescence (PL) quenching upon oxidation and PL recovery upon subsequent reduction. Time-resolved PL spectroscopy reveals a decrease in exciton lifetime correlated to the PL quenching upon LSP evolution. Finally, we report on the dynamics of LSPs in nonstoichiometric copper chalcogenide NCs. Through pump-probe experiments, we determined the time constants for carrier-phonon scattering involved in LSP cooling. Our results demonstrate that copper chalcogenide NCs offer the unique property of holding excitons and highly tunable LSPs on demand, and hence they are envisaged as a unique platform for the evaluation of exciton/LSP interactions. |
| [117] |
Ren K, Yin P F, Zhou Y Z, Cao X Z, Dong C K, Cui L, Liu H, Du X W . Small, 2017,13(36):1700867.
|
| [118] |
Zhu D X, Tang A W, Kong Q H, Zeng B, Yang C H, Teng F . J. Phys. Chem. C, 2017,121(29):15922.
|
| [119] |
Shu Q W, Yang M J . Alloys Compd., 2016,660:361.
|
| [120] |
Van Der Stam W, Berends A C, Rabouw FT, Willhammar T, Ke X, Meeldijk J D, Bals S, De Mello Donega C . Chem. Mater., 2015,27(2):621.
|
| [121] |
Lee S, Baek S, Park J P, Park J H, Hwang D Y, Kwak S K ,Kim S W. Chem. Mater, 2016,28(10):3337.
|
| [122] |
Maiti P S, Ganai A K, Bar-Ziv R, Enyashin A N, Houben L ,Bar Sadan M. Chem. Mater, 2018,30(14):4489.
|
| [123] |
Niezgoda J S, Yap E, Keene J D, Mcbride J R ,Rosenthal S J. Nano Lett, 2014,14(6):3262.
|
| [124] |
Su Z H, Sun K W, Han Z L, Liu F Y, Lai Y Q, Li J, Liu Y X . Mater. Chem., 2012,22(32):16346.
|
| [125] |
Li Y W, Ling W D, Han Q F, Kim T W, Shi W Z . Alloys Compd., 2015,633:347.
|
| [126] |
Lu X T, Zhuang Z B, Peng Q ,Li Y D . Chem. Commun, 2011,47(11):3141.
|
| [127] |
Li M, Zhou W-H, Guo J, Zhou Y L, Hou Z L, Jiao J, Zhou Z J, Du Z L, Wu S X . J. Phys. Chem. C, 2012,116(50):26507.
|
| [128] |
Lou Z Z, Gu Q, Xu L, Liao Y S, Xue C . Chem.-Asian J., 2015,10(6):1291.
|
| [129] |
Lou Z Z, Xue C . CrystEngComm, 2016,18(43):8406.
|
| [130] |
Pan L, Zhang J W, Jia X, Ma Y H, Zhang X W, Wang L ,Zou J J. Chin. J. Catal, 2017,38(2):253.
|
| [131] |
Lou Z Z, Zhu M S, Yang X G, Zhang Y, Whangbo M H, Li B J ,Huang B B. Appl. Catal. B: Environ., 2018,226:10.
|
| [132] |
Tan X J, Wang L Z, Cheng C, Yan X F, Shen B ,Zhang J L. Chem. Commun, 2016,52(14):2893.
|
| [133] |
Cheng H F, Kamegawa T, Mori K, Yamashita H . Angew. Chem 2014,126(11):2954.
|
| [134] |
Yin H B, Kuwahara Y, Mori K, Cheng H F, Wen M C, Yamashita H . J. Mater. Chem. A, 2017,5(19):8946.
|
| [135] |
Greenberg B L, Ganguly S, Held J T, Kramer N J, Mkhoyan K A, Aydil E S ,Kortshagen U R. Nano Lett, 2015,15(12):8162.
|
| [136] |
Cheng H F, Wen M C, Ma X C, Kuwahara Y, Mori K, Dai Y, Huang B B, Yamashita H . Am. Chem. Soc., 2016,138(29):9316. https://www.ncbi.nlm.nih.gov/pubmed/27384437
DOI: 10.1021/jacs.6b05396 PMID: 27384437 Heavily doped semiconductors have recently emerged as a remarkable class of plasmonic alternative to conventional noble metals; however, controlled manipulation of their surface plasmon bands toward short wavelengths, especially in the visible light spectrum, still remains a challenge. Here we demonstrate that hydrogen doped given MoO3 and WO3 via a facile H-spillover approach, namely, hydrogen bronzes, exhibit strong localized surface plasmon resonances in the visible light region. Through variation of their stoichiometric compositions, tunable plasmon resonances could be observed in a wide range, which hinge upon the reduction temperatures, metal species, the nature and the size of metal oxide supports in the synthetic H2 reduction process as well as oxidation treatment in the postsynthetic process. Density functional theory calculations unravel that the intercalation of hydrogen atoms into the given host structures yields appreciable delocalized electrons, enabling their plasmonic properties. The plasmonic hybrids show potentials in heterogeneous catalysis, in which visible light irradiation enhanced catalytic performance toward p-nitrophenol reduction relative to dark condition. Our findings provide direct evidence for achieving plasmon resonances in hydrogen doped metal oxide semiconductors, and may allow large-scale applications with low-price and earth-abundant elements. |
| [137] |
Milla M J, Barho F, González-Posada F, Cerutti L, Bomers M, Rodriguez J B, Tournié E, Taliercio T . Nanotechnology, 2016,27(42):425201.
|
| [138] |
Chen L, Sun H H, Zhao Y J, Zhang Y, Wang Y X, Liu Y, Zhang X, Jiang Y H, Hua Z ,Yang J H. RSC Adv, 2017,7(27):16553.
|
| [139] |
Himstedt R, Rusch P, Hinrichs D, Kodanek T, Lauth J, Kinge S ,Siebbeles L D A, Dorfs D . Chem. Mater., 2017,29(17):7371.
|
| [140] |
Wang Z L, Quan X J, Zhang Z M, Cheng P . Quant. Spectrosc. Radiat. Transf., 2018,205:291.
|
| [141] |
Wang H J, Yang K H, Hsu S C, Huang M H . Nanoscale, 2016,8(2):965.
|
| [142] |
Ghodselahi T, Vesaghi M A . Physica B, 2011,406(13):2678.
|
| [143] |
Seh Z W, Liu S H, Low M, Zhang S Y, Liu Z L, Mlayah A ,Han M Y. Adv. Mater, 2012,24(17):2310.
|
| [144] |
Yao G Y, Liu Q L, Zhao Z Y . Catalysts, 2018,8(6):236.
|
| [145] |
Takahata R, Yamazoe S, Koyasu K, Imura K, Tsukuda T . Am. Chem. Soc., 2018,140(21):6640. https://www.ncbi.nlm.nih.gov/pubmed/29694041
DOI: 10.1021/jacs.8b02884 PMID: 29694041 We synthesized gold ultrathin nanorods (AuUNRs) by slow reductions of gold(I) in the presence of oleylamine (OA) as a surfactant. Transmission electron microscopy revealed that the lengths of AuUNRs were tuned in the range of 5-20 nm while keeping the diameter constant (∼2 nm) by changing the relative concentration of OA and Au(I). It is proposed on the basis of time-resolved optical spectroscopy that AuUNRs are formed via the formation of small (<2 nm) Au spherical clusters followed by their one-dimensional attachment in OA micelles. The surfactant OA on AuUNRs was successfully replaced with glutathionate or dodecanethiolate by the ligand exchange approach. Optical extinction spectroscopy on a series of AuUNRs with different aspect ratios (ARs) revealed a single intense extinction band in the near-IR (NIR) region due to the longitudinal localized surface plasmon resonance (LSPR), the peak position of which is red-shifted with the AR. The NIR bands of AuUNRs with AR < 5 were blue-shifted upon the ligand exchange from OA to thiolates, in sharp contrast to the red shift observed in the conventional Au nanorods and nanospheres (diameter >10 nm). This behavior suggests that the NIR bands of thiolate-protected AuUNRs with AR < 5 are not plasmonic in nature, but are associated with a single-electron excitation between quantized states. The LSPR band was attenuated by thiolate passivation that can be explained by the direct decay of plasmons into an interfacial charge transfer state (chemical interface damping). The LSPR wavelengths of AuUNRs are remarkably longer than those of the conventional AuNRs with the same AR, demonstrating that the miniaturization of the diameter to below ∼2 nm significantly affects the optical response. The red shift of the LSPR band can be ascribed to the increase in the effective mass of electrons in AuUNRs. |
| [146] |
Mu H W, Lv J W, Liu C, Sun T, Chu P K ,Zhang J P. Opt. Commun, 2017,402:216.
|
| [147] |
Han C, Quan Q, Chen H M, Sun Y, Xu Y J . Small, 2017,13(14):1602947.
|
| [148] |
Hong J W, Wi D H, Lee S U, Han S W . Am. Chem. Soc., 2016,138(48):15766. https://www.ncbi.nlm.nih.gov/pubmed/27933998
DOI: 10.1021/jacs.6b10288 PMID: 27933998 2O on desired sites of anisotropic Au nanocrystals. Both the exploitation of structural characteristics of Au nanocrystals and the selective stabilization of their surfaces are keys to the construction of heteronanocrystals with a specific configuration. Our approach can provide an opportunity to precisely explore the link between the solar energy conversion efficiency and the structure of heteronanocrystals as well as to obtain important insights into the underpinning mechanism. Heteronanocrystals produced by Cu2O overgrowth preferentially on the multiple high-curvature sites of Au nanocrystals exhibited prominent photocatalytic hydrogen production activity due to efficient charge separation by strong plasmon excitation at the Au-Cu2O interface and subsequent sustainable hot electron transfer from Au to Cu2O.]]> |
/
| 〈 |
|
〉 |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}