






1 引言
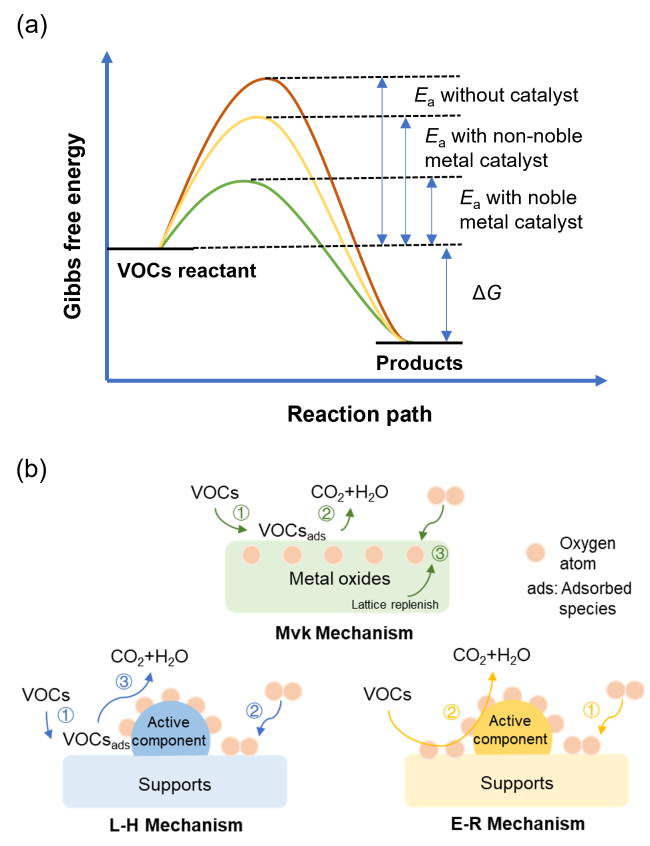
2 VOCs催化氧化机理
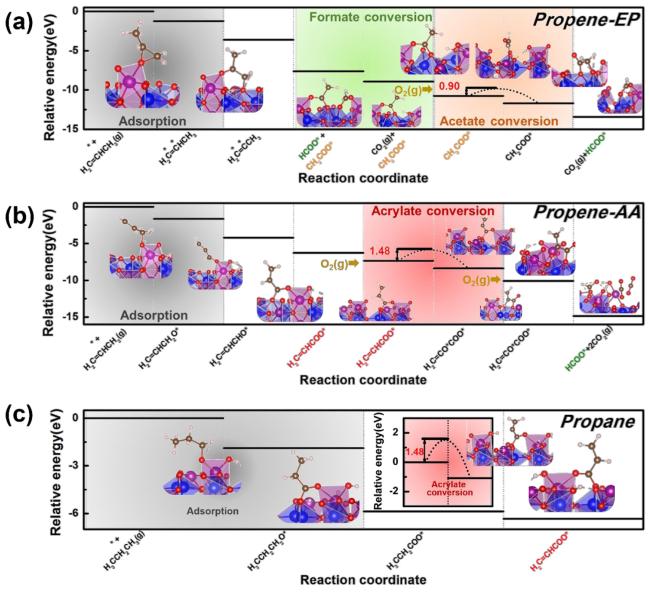
3 非贵金属催化剂结构调控
3.1 单一过渡金属氧化物
3.1.1 氧化态与晶型
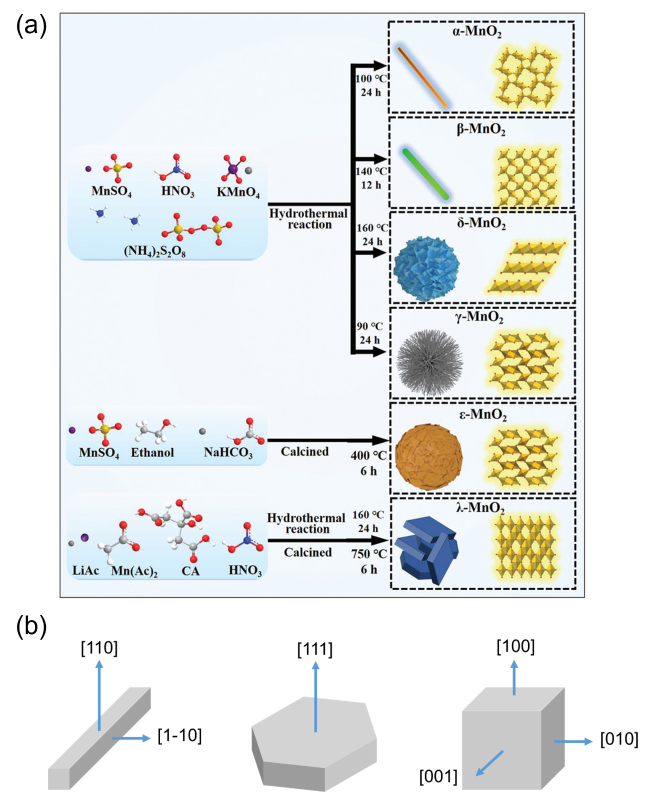
3.1.2 形貌与暴露晶面
3.2 混合金属氧化物
3.2.1 元素掺杂
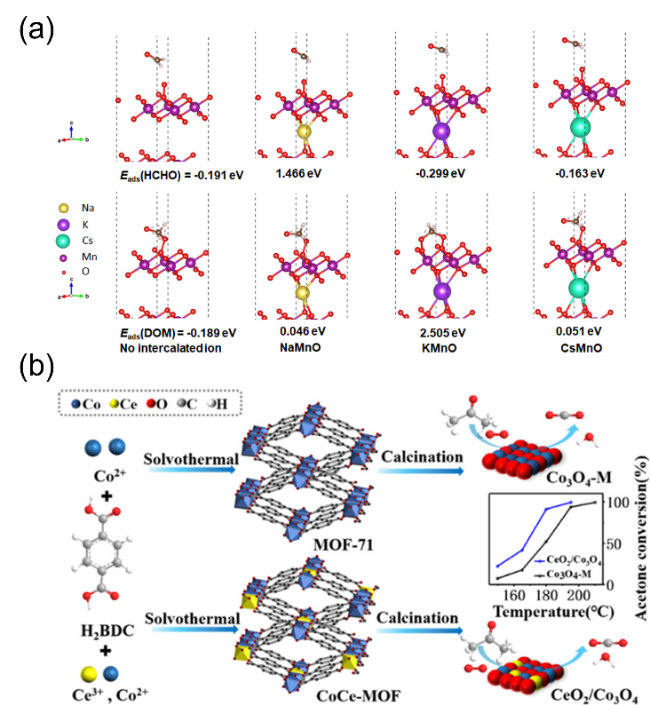
图4 (a)δ-MnO2不同层间离子掺杂对甲醛及其氧化中间物种吸附能的影响[43],(b)MOF-71衍生Co-Ce混合氧化物及其丙酮氧化活性[73]Fig. 4 (a) The effect of different interlayer ions in δ-MnO2 on the adsorption energy of formaldehyde and oxidation intermediate species[43], Copyright 2020, American Chemical Society; (b) MOF-71-derived Co-Ce mixed oxides and their acetone oxidation activity[73], Copyright 2020, American Chemical Society |
3.2.2 固溶体
3.2.3 特殊前驱体调控
3.3 复合金属氧化物
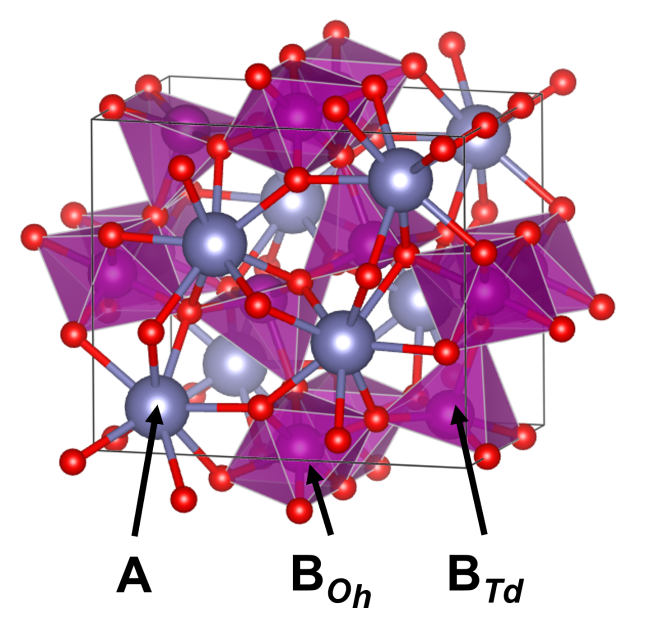
3.3.1 尖晶石
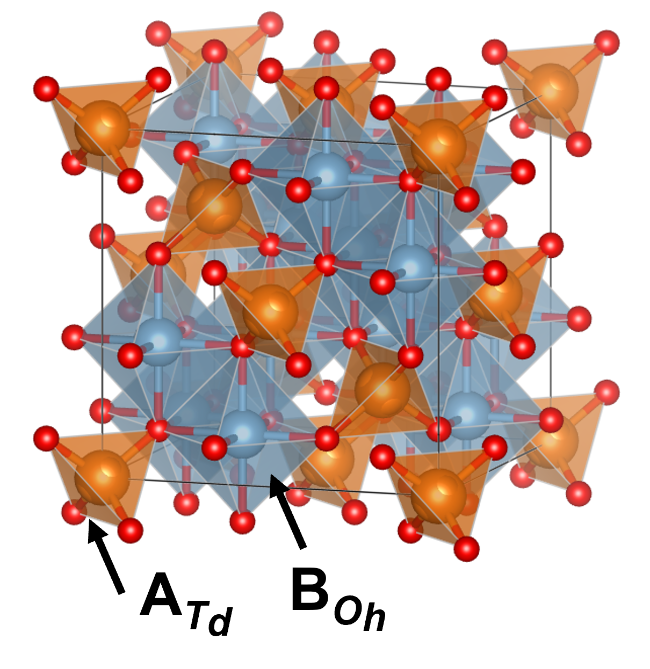
3.3.2 钙钛矿
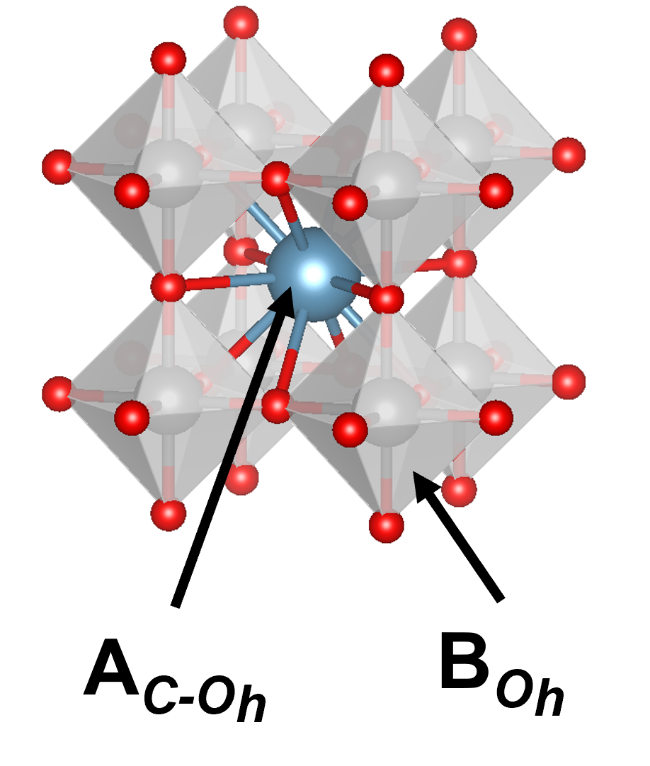
3.3.3 莫来石
3.4 相界面结构调控
3.4.1 负载型异质结构
3.4.2 核壳型异质结构
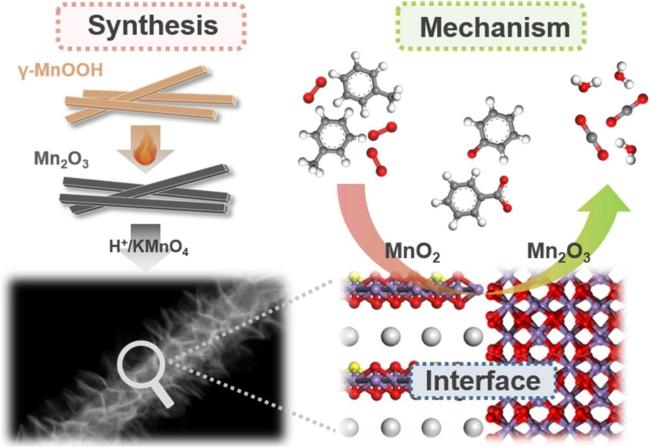
4 贵金属催化剂贵金属分散状态调控
4.1 贵金属纳米颗粒/团簇催化剂
4.1.1 尺寸效应
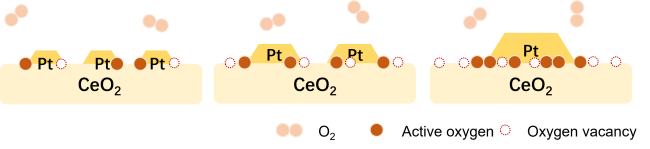
4.1.2 载体效应

图10 (a)Pd负载于不同形貌CeO2表面时其CO与丙烷氧化反应速率[152],(b)MOF原位生长诱导Co3O4与Pt NPs间电子转移促进甲苯氧化机理示意图[153]Fig. 10 (a) The reaction rate of CO and propane oxidation over Pd loaded on CeO2 with different morphologies[152], Copyright 2016, American Chemical Society; (b) Schematic diagram of electron transfer between Co3O4 and Pt NPs induced by MOF in situ growth promoting toluene oxidation[153], Copyright 2022, American Chemical Society |
4.2 贵金属单原子催化剂
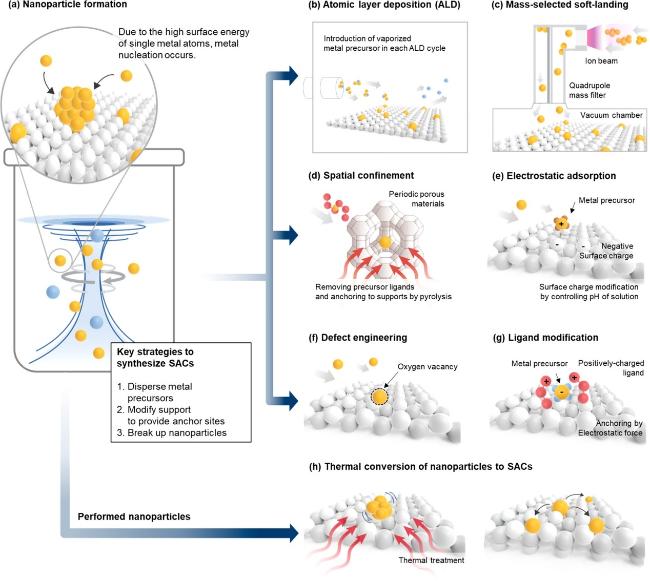
4.2.1 单原子催化剂的制备策略
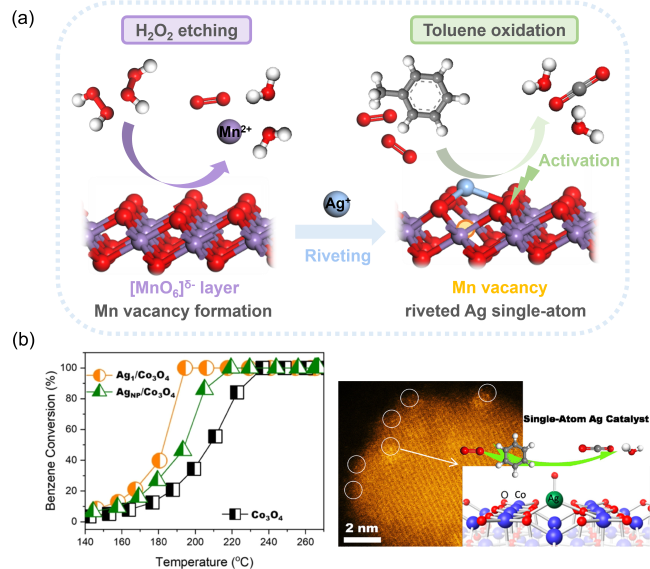
4.2.2 金属-载体相互作用调控
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
图12 (a)H2O2辅助MnO2表面Mn空位捕获Ag单原子促进氧活化[179];(b)Co3+位点稳定Ag单原子促进苯氧化[180]Fig. 12 (a) H2O2-assisted Mn vacancy capture of Ag single atom on MnO2 surface promoting oxygen activation[179], Copyright 2022, Royal Society of Chemistry; (b) Co3+-site stabilized Ag single atom promoting benzene oxidation activity[180], Copyright 2022, American Chemical Society |