



1 引言
2 低温下CO2氢化催化剂性能与转化途径
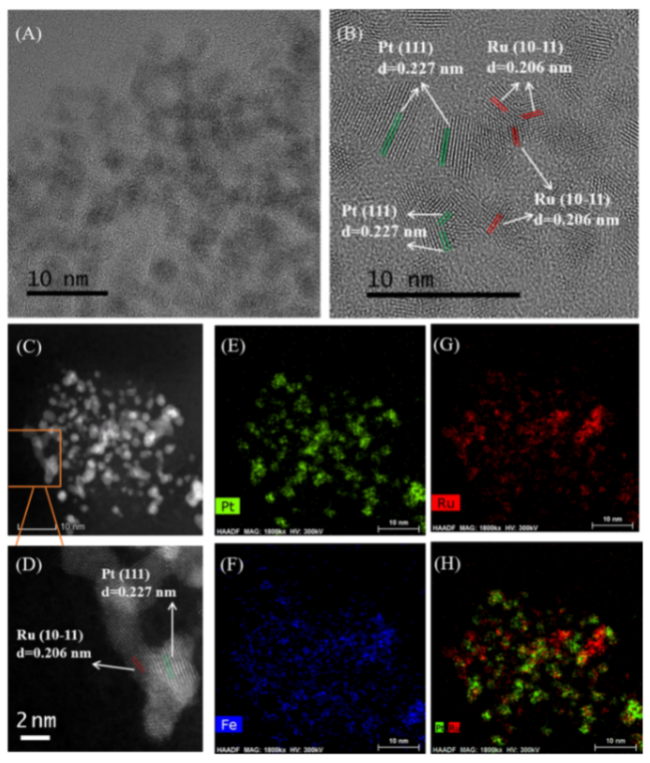
图1 (A~C) Ru-Pt/FeCO3的透射电镜照片、高分辨透射电镜照片和高角度环形暗场扫描透射电镜照片,(D) C图中选区高分辨照片,(E~G) 选区内Pt、Fe和Ru的元素分布图,(H) 图E和图G的合并图片[28]Fig.1 (A~C) TEM image, high-resolution TEM image, and HAADF-STEM image of Ru-Pt/FeCO3, (D) High-resolution TEM image of the selected area in (C), (E~G) EDX elemental mapping of Pt, Fe, and Ru in the same area, (H) Merged image of (E) and (G)[28]. Reproduced from Ref. 28 with permission from the Royal Society of Chemistry |
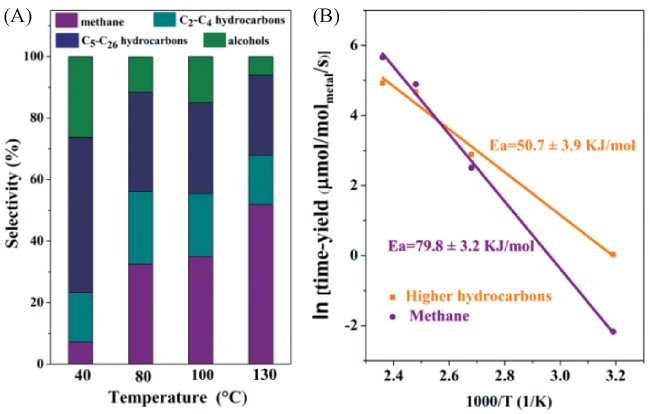
图2 (A)在不同温度下Ru-Pt/FeCO3催化CO2氢化产物分布图, (B)该催化体系中CO2氢化生成甲烷和多碳化合物的表观活化能[28]Fig.2 (A) Product distributions for CO2 hydrogenation over Ru-Pt/FeCO3 at different temperatures. (B) Apparent activation energy measurements for the formation of methane and higher hydrocarbons (C2+) in CO2 hydrogenation over Ru-Pt/FeCO3, respectively[28]. Reproduced from Ref. 28 with permission from the Royal Society of Chemistry |
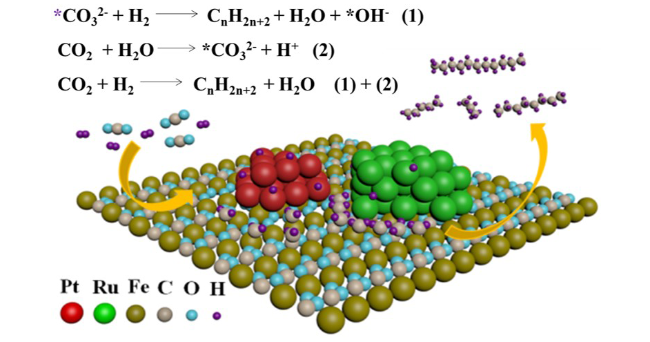
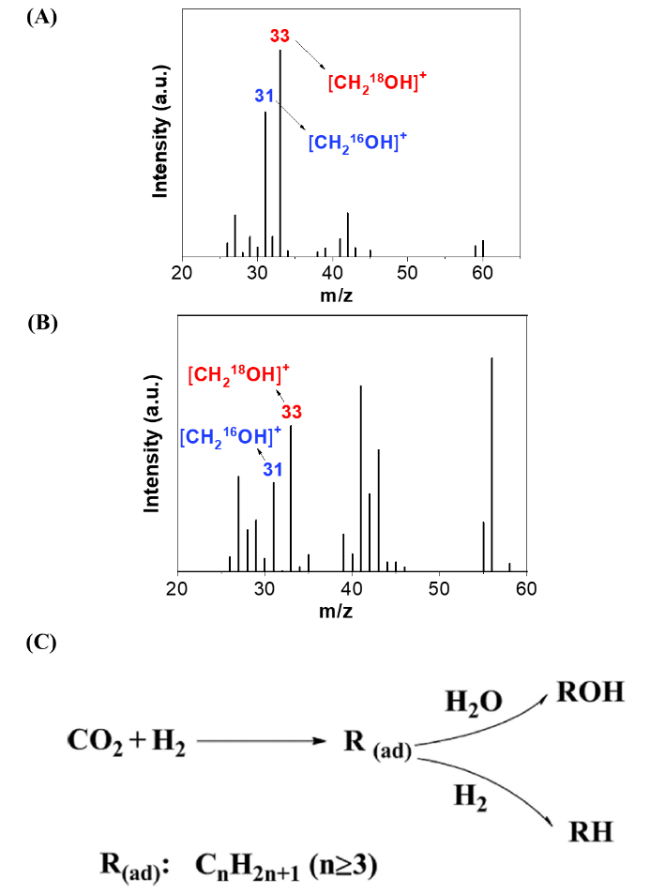
图4 (A, B)在18O标记水中Ru-Pt/Fe3O4催化CO2氢化反应生成的丙醇和丁醇的质谱图,(C)低温下金属烷基水解生成多碳醇的反应途径示意图[30]Fig.4 (A, B) The mass spectra of propanol and butanol formed in CO2 hydrogenation over Ru-Pt/Fe3O4 in18O labeled water, (C) reaction mechanism for multi-carbon alcohols formation based on metal-alkyl hydrolysis over Ru-Pt/Fe3O4[30] |
3 低温下CO2氢化产物选择性调控化学机制
4 高选择性PtRu双金属纳米簇催化剂的合成与性能

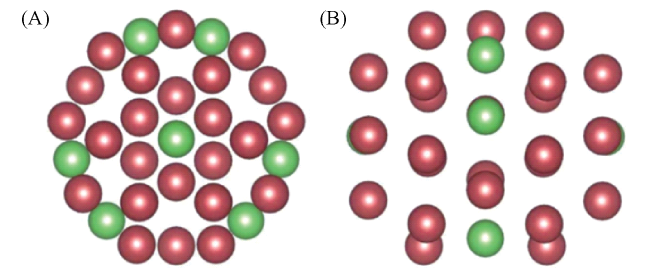
图6 (A) Ru-co-Pt/C的高角度环形暗场扫描透射电镜照片,(B~D) 选区中Pt、Ru以及Pt和Ru叠加的元素分布图像,(E) 选区中金属纳米粒子的原子级分辨图像,(F) Ru-co-Pt模型,其中红色球代表一列原子柱中主要含有Pt原子,绿色球代表一列原子柱中主要含有Ru原子,(G) E图中选区中粒子的放大图像,(H) 双金属纳米粒子中原子(或柱)Z-衬度线扫分析结果[31]Fig.6 (A) The HAADF-STEM image of Ru-co-Pt/C. (B~D) Energy dispersive X-ray spectroscopy (EDX) elemental mapping image of Pt (B), Ru (C), and Pt + Ru (D) in the selected area. (E) The atomic-resolution HADDF-STEM image of a typical particle in the selected area. (F) A model for the structure of Ru-co-Pt, in which the atom columns mainly composed of Pt and Ru are marked with red and green balls, respectively. (G) The enlarged HAADF-STEM image of the selected typical bimetallic particle in (E). (H) Line scan Z-contrast analysis of atom columns in the bimetallic nanoparticle in (E) along the arrow in (F)[31]. Reproduced from Ref. 31 with permission from the Royal Society of Chemistry |
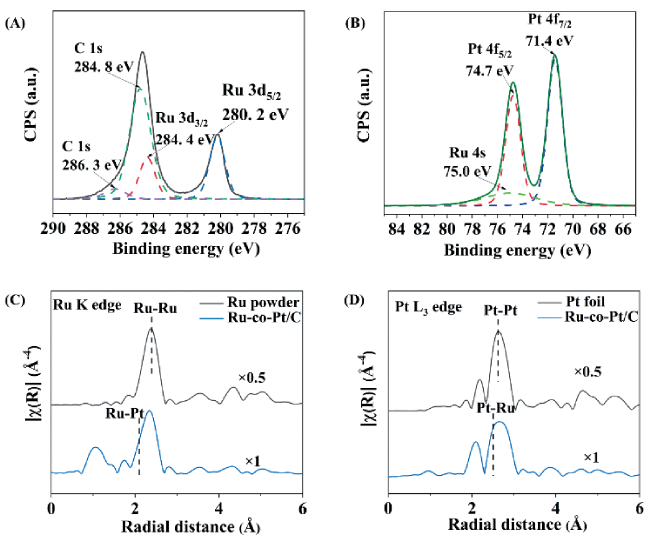
图7 (A,B) Ru-co-Pt/C中Ru和Pt的X射线光电子能谱图, (C, D) Ru-co-Pt/C样品的Ru K边和Pt L3边的扩展X射线吸收精细结构谱图[31]Fig.7 (A, B) XPS spectra of Ru 3d (A) and Pt 4f levels (B) in Ru-co-Pt/C, respectively. (C, D) Fourier transform (FT) EXAFS spectra of Ru K edge (C) and Pt L3 edge (D) for Ru-co-Pt/C, respectively[31]. Reproduced from Ref. 31 with permission from the Royal Society of Chemistry |
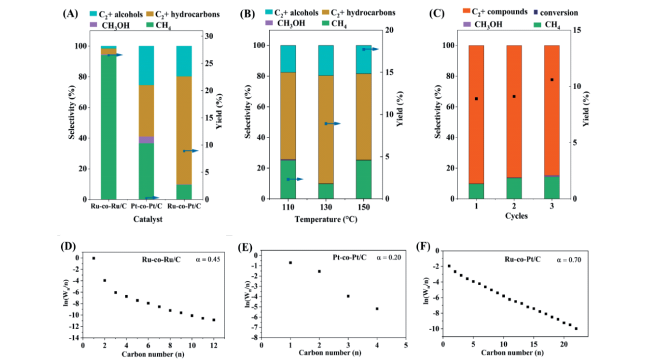
图8 (A)Ru-co-Ru/C、Pt-co-Pt/C和Ru-co-Pt/C三种催化剂催化CO2加氢反应性能,(B)Ru-co-Pt/C在不同温度下催化CO2加氢反应性能,(C)Ru-co-Pt/C在130℃下催化CO2加氢稳定性测试,(D~F)Ru-co-Ru/C、Pt-co-Pt/C和Ru-co-Pt/C催化CO2加氢生成的产物分布图,Wn代表Cn物质的质量分数[31]Fig.8 (A) Catalytic performance of Ru-co-Ru/C, Pt-co-Pt/C, and Ru-co-Pt/C in CO2 hydrogenation, respectively. (B) Catalytic performance of Ru-co-Pt/C in CO2 hydrogenation at different temperatures. (C) The stability of Ru-co-Pt/C in CO2 hydrogenation at 130℃. (D~F) Product distributions in CO2 hydrogenation over Ru-co-Ru/C (D), Pt-co-Pt/C (E), and Ru-co-Pt/C (F) at 130℃, respectively. Wn represents the mass percentage of Cn compounds in the products [31]. Reproduced from Ref. 31 with permission from the Royal Society of Chemistry |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
表1 由DFT理论计算获得的CO2氢化反应中若干基元反应能垒[31]Table 1 The energy barriers of several primitive reactions in CO2 hydrogenation calculated via DFT. [31] Reproduced from Ref. 31 with permission from the Royal Society of Chemistry. |
| Catalyst | Reaction energy barriers (eV) | ||
|---|---|---|---|
| CH2+CH2 coupling | CH2 + H | CH3 +H | |
| Pt42-Ru8 | 0.31 | 0.55 | 0.53 |
| Ru50 | 1.44 | 0.85 | 0.77 |
| Pt50 | 0.78 | 0.92 | 0.81 |