



1 引言
氮气分子是物质世界和生命物质中氮元素最重要的来源。氮气储量丰富、廉价易得,可谓是取之不尽、用之不竭的氮源。但是空气中游离的氮气无法被人类或者动植物直接吸收利用,只有将游离态的氮转变为化合态的氮(即氮的固定)才能被大部分生物所利用。发展高效的转化方法将氮气分子中的氮固定,变成各种各样功能的含氮化合物,长期以来是学术界和工业界的“圣杯”。然而众所周知,氮气分子的化学性质非常稳定,常温下很难与其他物质发生反应。因此,氮气分子的高效活化与转化是横亘一个世纪的科学难题。幸运的是,一百多年前人类就建立了由氮气和氢气合成氨的过程(Haber-Bosch合成氨工艺),实现了氨的规模化生产。基于合成氨技术,人类在应对粮食问题的同时,也为各种各样功能化合物分子提供了“可用的氮源”。换言之,目前氨是几乎所有含氮化合物的氮元素来源,工业合成氨是人类合成史上大规模转化氮气的唯一途径。但是,由于传统合成氨工业的高能耗、高污染等问题,以及将NH3转化成部分重要含氮化合物所面临的困难,化学家们一直在探索改进甚至颠覆工业合成氨技术,同时寻求开发直接利用氮气的其他途径。
在过去的一个多世纪里,科研工作者们分别从均相化学、多相化学、酶化学等不同角度对氮气转化过程进行了系统而深入的研究,获得了诸多新认识,形成了许多新概念与新方法,合成了一系列新化合物与新材料,极大地丰富了固氮知识体系。尽管人们已取得了这些重要的研究进展,但距离实现“氮气的可控转化”这一宏伟目标仍有很长的路要走。部分原因归因于对氮气转化这一科学问题的认识是学科化的、片面的,而缺乏整体的、全面的认识。
最近,吉林大学徐如人先生等提出了“凝聚态化学”的概念。凝聚态是由具有特定电子结构且在传统化学上被视为反应主体的原子、离子以及由化学键(离子键、共价键、金属键、配位键、氢键)键合的分子等“基本粒子”,凭借电相互作用(库仑相互作用、电子轨道杂化和叠加等)建立“稳定黏连关系”而形成,具有特定组成、多层次结构、性质与功能[1⇓~3]。以晶体固态为例,凝聚态的结构主要可以概括为三个层次:第一层次是组成固态的基本粒子的电子结构和几何结构;第二层次是组成固态的晶体结构;第三层次是固态宏观结构。徐先生等指出,在实际反应中,凝聚态物质才是所有化学反应的真实主体。在发生化学反应时,物质的凝聚态结构也将发生变化。因此,仅从传统分子与理想晶体模型来理解固氮化学是不够深刻和全面的。凝聚态化学为固氮反应化学的深入认识提供了方向和基础。本文从凝聚态化学的角度,在均相溶液体系固氮、氮气/氧气多能耦合转化、多相合成氨三大方面,探讨了固氮化学领域的一些科学问题,期望本综述可以为固氮化学研究提供有益参考。
2 均相溶液体系固氮中的凝聚态化学
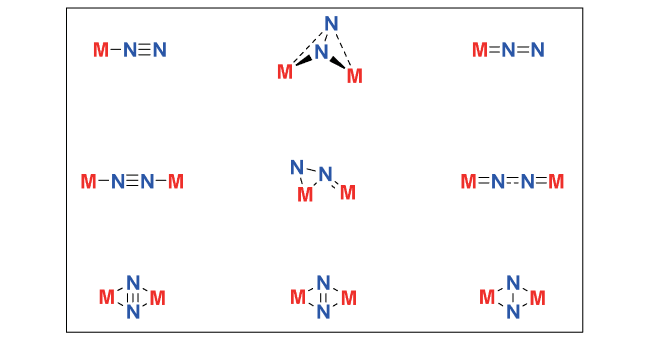
为了实现温和条件下均相溶液体系中的氮气活化与转化,科学家们进行了大量探索。在溶液体系中,实现氮气活化的重要途径是利用过渡金属与氮气形成配合物[4⇓⇓⇓⇓⇓⇓⇓~12]。1965年,Senoff等[13]合成了第一例过渡金属-氮气配合物[Ru(NH3)5N2]X2 (X = Br-, I-, B , P );1968年,Ibers等[14]首次报道了过渡金属-氮气配合物CoH(N2)(PPh3)3的晶体结构。经过国内外化学家的不懈努力,截至2020年3月,超过800例具有明确结构的金属-氮气配合物被英国剑桥晶体数据中心(Cambridge Crystallographic Data Centre, CCDC)收录,涵盖了丰富多彩的金属-氮气配位模式(图1 )。但总的来说,表征明晰的结构数量有限,且仅有为数不多的金属-氮气配合物能够发生氮气衍生化反应[15⇓⇓⇓⇓~20]。
目前通过金属氮气配合物实现氮气衍生化反应研究较为成功的例子,主要集中在被活化的氮气与质子的反应(成氨反应)上。1975年,Chatt[21]报道了零价Mo和W的氮气配合物与酸反应,当量生成氨的过程,并提出了金属-末端氮气配合物逐步质子化释放氨的机理,即“Chatt循环”。2003年,Yandulov和Schrock等[22]报道了首例均相合成氨催化剂Mo[(HIPTN)3N](N2),该配合物可以在外加质子({2,6-lutidinium}{BAr'4}, Ar' = 3,5-(CF3)2C6H3)和电子供体(CrC )的条件下实现常温常压N2的催化转化,即“Schrock循环”。2010年,Nishibayashi等[23]报道了PNP配体支持的双核Mo基配合物[Mo(N2)2(PNP)]2(μ-N2),通过使用CoCp2作为还原剂,实现了常温常压下氨的催化合成,并大幅提高了产氨效率。2013年,Peters等[24]成功将催化剂由Mo体系拓展到Fe体系,报道了Fe-N2配合物催化剂[(TPB)Fe(N2)][Na(12-crown-4)2],使用强还原剂石墨钾作为电子供体,在低温反应条件下实现了催化产氨循环。
这些里程碑式合成氨体系的发现,离不开对关键中间体的详细表征和对反应机理的深入探索。在这个过程中,科学家逐渐认识到,氮气的活化强烈地受中心金属氧化态、配位环境和配位模式等因素的影响,微小的改变可能带来截然不同的结果。Dewar-Chatt-Duncanson(DCD)理论模型可以部分解释该配位模式下氮气的活化模式。如图2 所示,过渡金属的占据d电子可以与氮气分子的π*轨道形成反馈π键,氮气孤对电子则与过渡金属空的d轨道形成σ配位键,非极性的氮气分子被一定程度极化,末端Nβ原子将携带部分负电荷。氮气在此过程中,从金属中心得到电子,被一定程度还原。
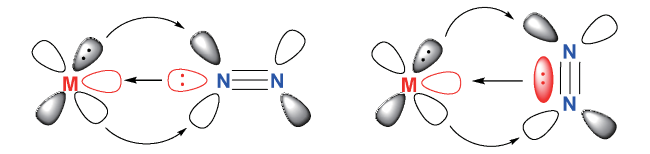
DCD理论模型揭示了氮气活化的基本原理,但依然有较大局限性。因为溶液体系中,金属配合物往往不是以单体的形式存在,而是会形成复杂的多聚簇合物。氮气从气相进入液相,进而与金属团簇进行配位,最后形成稳定的金属氮化物的过程中,会涉及复杂的凝聚态过程。同时,均相溶液体系固氮过程中会经常使用不溶于溶剂的固体还原剂,如石墨钾、钠汞齐、金属镁、金属钾等,在还原反应发生的过程中,往往还会伴随着固体盐产物的析出。这些金属团簇(溶液相)与氮气(气相)和还原剂(固相)分处于三个不同的物相中,它们之间的相互作用,不能简单地通过单中心金属-氮气的配位模式来进行分析。另外,反应溶剂、还原剂、添加剂甚至体系中原位生成的金属盐又可能反过来影响溶液中金属团簇的状态,很大程度地影响反应结果。相应的例子很多,限于篇幅,本文在此仅举几例进行说明。
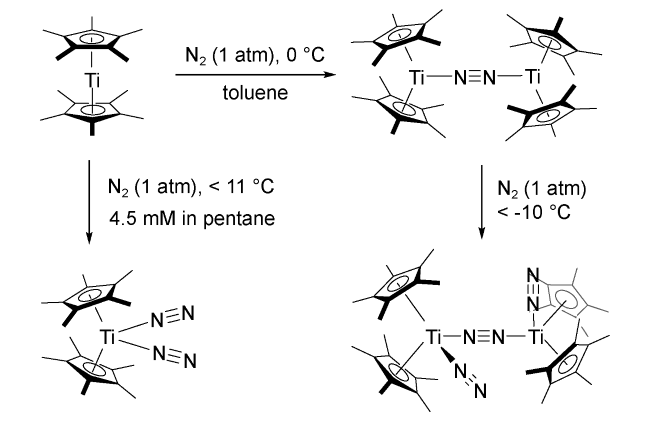
以茂基金属钛-氮气配合物为例(图式1 )。Bercaw等[25]在1971年报道了五甲基取代的二茂钛溶液可以在常压下与氮气反应,形成摩尔比2∶1的金属配合物。在随后的系列工作中发现,溶液状态下的Ti-N2物种形态强烈地受到溶剂、氮气压力、温度以及Ti配合物浓度等因素的影响。例如,直线型Ti-NN-Ti配合物是一种蓝黑色晶体,可以在甲苯中0℃下结晶分离得到[26]。但是如果在一个氮气压力下,继续降低体系的温度(<-10℃),每个Ti原子上会再额外配位一个氮气分子,形成蓝紫色的晶体[27]。然而如果降低溶液体系中二茂钛的浓度,同时更换溶剂为正戊烷,使之可以溶解相对更多的氮气,那么就可以分离得到单核茂基氮气配合物[28]。
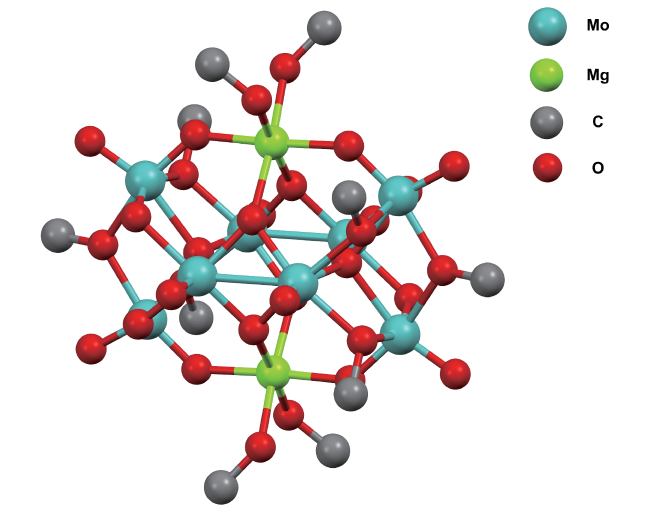
不仅是Ti配合物和氮气反应时会表现出如此丰富的凝聚态,在其他过渡金属-氮气配合物体系中,也能观察到类似的现象。Shilova等[31]在早期研究过渡金属盐活化氮气时发现,如果使用合适的混合金属盐作为催化剂,其催化成氨或肼的效率相较于使用单一金属盐会明显提高。例如,MoCl5和MgCl2在碱性条件下混合于甲醇溶液中可以观察到多种结构非常复杂的Mo-Mg簇合物,其中被分离鉴定的一种簇合物为[Mg2Mo8O22(OMe)6(MeOH)4][Mg(MeOH)6](MeOH)6 (图3 )。它可以在钠汞齐和磷脂存在下,于甲醇溶液中催化氮气转化为肼,且活性良好。值得一提的是,溶液中还存在其他多种不同核数的Mo-Mg凝聚态结构,这些未被深入探索的复杂结构很可能表现出不同的氮气活化效果[32]。当使用不同的金属盐组合,或者将桥连基团甲醇(甲氧基)更换为其他基团时,溶液中簇合物凝聚态的结构会如何变化呢?这是一个值得深入探究的问题。
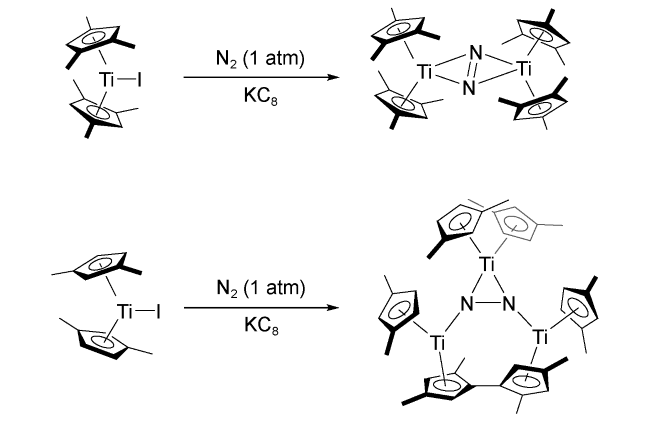
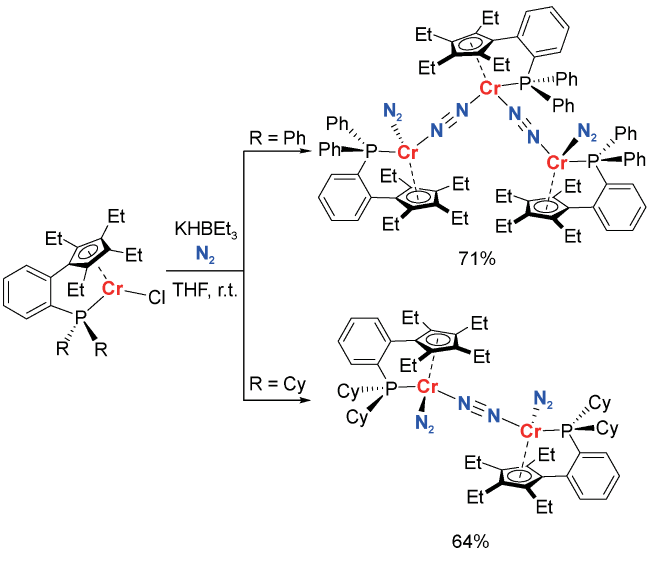
另一个例子是席振峰课题组[33]报道的金属铬氮气配合物的氮衍生化反应(图式3 )。席振峰课题组利用其开发的环戊二烯-有机膦配体(L1配体),有效稳定铬金属中心,得到了相应的L1-CrCl配合物。在常温、常压氮气下,使用三乙基硼氢化钾(KHBEt3)或石墨钾(KC8)还原该二价铬氯化物,可以得到一价铬氮气配合物。更换环戊二烯-有机膦配体上取代基,会得到复杂的双核或三核的一价铬氮气配合物,其中一价铬氮气配合物的金属铬中心配位氮气的数量还可以不同,形成对称结构或者不对称结构。在这些簇合物凝聚态结构中,处于不同位置的氮气与金属中心的结合模式不同,因此被活化程度也不相同。但这样配位模式的氮气,都不易发生进一步的氮气衍生化反应。
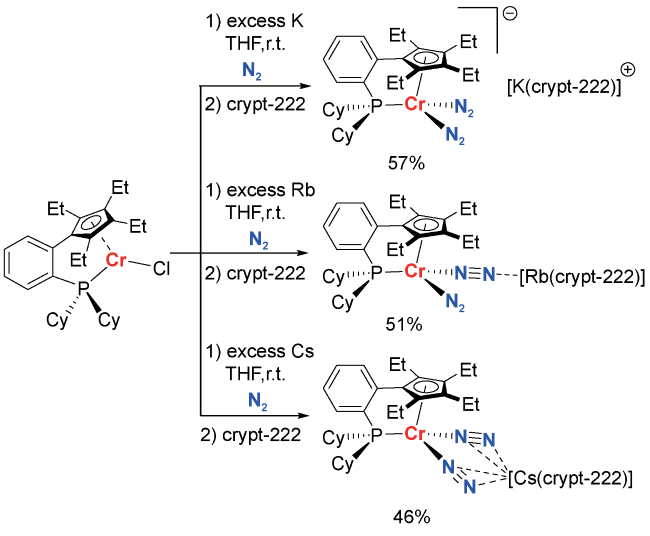
为了实现配位氮气的衍生化,需要进一步降低金属中心的氧化态(图式4 )。一价铬氮气配合可以被过量的碱金属(钾、铷、铯)继续还原,得到电子,生成相应的零价铬氮气配合物。晶体结构显示,碱金属被穴醚-[2.2.2]所络合,每个零价铬中心配位有两个氮气分子。但是根据金属抗衡离子种类的不同,钾、铷和铯同样作为路易斯酸,与氮气的距离和结合方式却有明显不同。其对氮气活化程度的影响,通过红外光谱可以得到证实。
尽管过渡金属活化氮气的理论模型早已构建,相应配合物的晶体结构也有相当积累,但是人们对金属配合物不同聚集状态对氮气活化影响的理解仍然非常有限,相关详细的表征数据非常匮乏。特别的,目前均相体系活化氮气的研究主要是在一个大气压下进行的,在相对高压(10~200个大气压)时,真实溶液体系中金属团簇与氮气的结合数量、结合模式以及活化方式等均是未知。另外,单晶结构与溶液体系中的真实活性物种并不在所有情况下都能完全对应,特别是在体系中存在添加剂、盐以及配位溶剂时,真实的金属-氮气活性物种的凝聚态结构可能会非常复杂,与高度简化的理论模型相去甚远。最后,从氮气转变为含氮有机物是一个多步过程,尤其是其中伴随着复杂的从还原剂(固相)到氮气配合物(溶液相)的电子转移过程,随着氮气的衍生化反应的进行,溶液中形成的关键中间体的凝聚态结构很可能无法保持不变,会发生复杂的变化。后续相关领域的化学家,需要发展更多监测和表征溶液中活性中间体的凝聚态真实结构的手段。通过对这些凝聚态的系统探索,有助于深刻理解实际体系下氮气的真实活化方式,为设计后续氮气的衍生化反应提供有益指导。
3 多相合成氨中的凝聚态化学
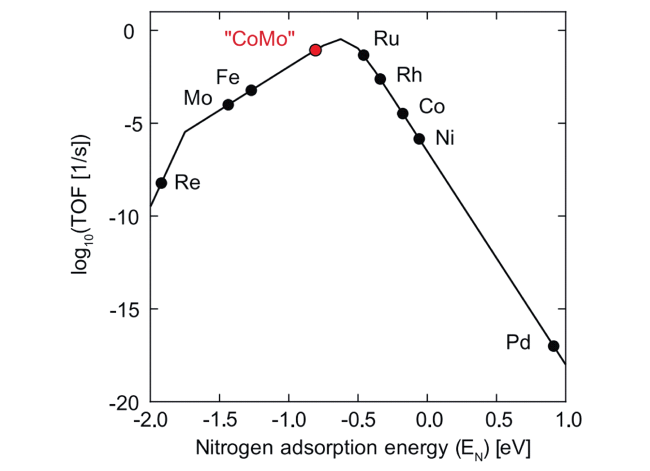
氮气(N2)和氢气(H2)在固体催化剂作用下生成氨(NH3)的反应,是典型的凝聚态反应过程。凝聚态的多层次结构对合成氨反应历程及动力学有着显著影响。与其他多相催化反应过程一样,合成氨亦需经历反应物(气态氮气和氢气)吸附、表面反应和产物(气态氨)脱附等步骤。固体催化剂表面若干原子形成的活性位点主导着这一化学过程的反应速率。过渡金属是合成氨催化剂的重要组分。一般认为,在Fe、Ru等过渡金属多相催化剂体系中,尤其是高温条件下直接解离式机理占主导[34⇓~36]。N2和H2在过渡金属表面分别发生吸附解离生成N原子(Nad)和H原子(Had),随后N原子逐步加氢生成N (x=0~3)物种,最后NH3从催化剂表面脱附(图4A )。合成氨总包反应速率与各基元步骤的活化能以及指前因子相关。其中,N2分子的解离化学吸附通常被认为是过渡金属表面合成氨反应的速率控制步骤。Ertl[37,38]采用表面科学技术考察了金属Fe表面N2、H2、NH3的吸附以及中间物种的生成与转化过程,构建了Fe表面上合成氨反应的位能图,从原子层面上较为清晰地揭示了该反应的微观历程(图4B )。通过大量科学研究,人们发现合成氨反应速率与过渡金属表面的氮吸附能之间存在着火山型曲线关系。这一现象可以从经典的萨巴蒂埃(Sabatier)原理或者近期由Nørskov等[39]提出的能量限制关系(Scaling relations)等进行理解(图5 )。
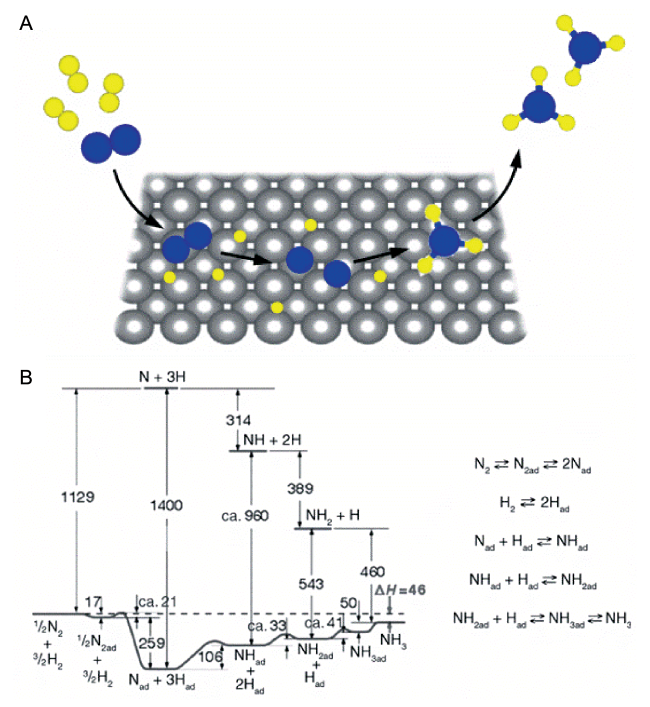
图4 (A)固体催化剂表面合成氨反应的直接解离式机理示意图;(B)金属Fe表面上合成氨反应势能图,能量单位为kJ·mol-1[37]Fig.4 (A) Schematic representation of ammonia synthesis on a solid surface via the dissociative mechanism; (B) potential energy diagram of ammonia synthesis on Fe surface. The energies are given in kJ·mol-1. Reprinted with permission[37]. Copyright John Wiley and Sons |
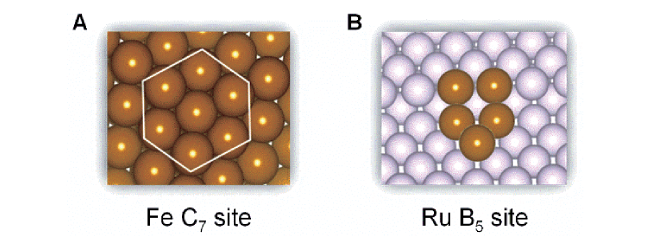
固体表面通常是非常复杂且不均匀的。迄今为止,我们对于气固相表界面的组成、结构及微观反应过程仍然缺乏足够的认识。如何理解活性位的本质是多相催化中最为基础的科学问题。关于过渡金属表面上合成氨反应的活性中心结构,研究人员基于大量表面科学实验及理论计算研究提出了可能的模型。目前一般认为,过渡金属上N2的活化位点由其表面上多个相邻的原子组成,如Fe基催化剂的活性位由7个相邻的Fe原子构成(C7位,图6A )[42,43],Ru的活性位则是由位于台阶处的5个相邻Ru原子所组成(即B5位,图6B )[44,45]。根据Wulff规则,尺寸为2 nm的Ru纳米颗粒具有最多的B5位数目,因而将具有较高的合成氨催化活性。然而随着材料合成与表征技术的快速发展,这一传统认识受到了挑战。例如,近期Li等[46]制备了金属有机框架材料负载的Ru亚纳米团簇催化剂(Ru/MIL-101,Ru粒径在1 nm左右),其合成氨活性反而优于Ru粒径为2~4 nm的负载型Ru基催化剂(如Ru/MgO等)。作者认为这是由于在实际反应条件下,Ru亚纳米团簇发生表面重构生成了较多B5活性位。Jiang等[47]考察了Ru的粒径大小对合成氨性能的影响,发现尺寸更小的Ru团簇较Ru纳米颗粒表现出更高的TOF值。过渡金属团簇物种有可能难以实施N≡N键的断裂,在这种情况下,氢助解离式合成氨路径更为可行。Li等[48,49]利用DFT计算构建了Fe3和Rh1Co3表面团簇活性中心,发现N2发生直接解离的能垒较高,而更倾向于加氢生成N2H物种。
为了提高过渡金属的原子利用率,通常需要将活性金属负载在某种载体上。对于某些功能性载体而言(如电子化合物、可还原性稀土氧化物等),载体不仅起到分散活性金属的作用,还可通过金属-载体间的相互作用对过渡金属的几何结构与电子性质产生影响,继而提升合成氨反应速率。如,Hosono等报道了一类具有较低功函的无机电子化合物载体(如[Ca24Al28O64]4+: 4e-和[Y5Si3]0.79+: 0.79e-等),这些化合物的强给电子能力增强了Ru的d电子对N2反键轨道的反馈作用,从而进一步削弱了N≡N键,加快了N2的解离吸附[51⇓~53]。Nagaoka等[54,55]发现经过高温预还原的稀土氧化物(如La0.5Ce0.5O1.75、La0.5Pr0.5O1.75等)在合成氨反应条件下能够与金属Ru产生强的金属-载体相互作用,从而使得Ru的电子云密度得到增强,促进N2的解离吸附。Chen等[56]则发现在合成氨反应条件下,稀土氧化物载体如Sm2O3表面上原位产生的活性Sm-H物种可以有效提高Ru团簇表面电子密度,促进N2的活化解离。而且作为一种活性氢物种,其可以直接参与反应过程中氨的生成,显著提高合成氨活性。
在合成氨反应中,助剂通常是催化剂的关键构成组分。在合成氨工业实践中,人们很早就认识到助剂如碱(土)金属的添加可显著提升过渡金属的催化性能。然而对于助剂的化学状态及其作用机制一直存在着争议。Ozaki、Ertl、Aika等[57⇓~59]认为碱(土)金属具有很强的给电子能力,其可以作为电子供体将电子转移给过渡金属,降低过渡金属表面功函,增强过渡金属与N2之间的电子授受作用,从而削弱N≡N键,促进N2的活化解离;而Somorjai等[60]认为碱金属的存在可以加速产物NH3的脱附,从而使过渡金属暴露出更多的位点用于氮气活化;Nørskov等[61]则认为碱金属促进效应的产生主要源于碱金属与反应中间态或过渡态物种之间的静电相互作用。近期,Nørskov等[62]又提出对于磁性金属(如Co、Ni)催化剂而言,碱(土)金属助剂的加入亦能够有效降低磁性金属表面的自旋极化效应,从而进一步降低N2的解离能垒。除作为电子助剂外,碱(土)金属亦可作为催化剂的关键活性组分参与合成氨反应。Chen等[63]发现通过向过渡金属中引入碱(土)金属氢化物,可以强烈改变活性金属的催化行为。如氢化锂(LiH)的引入,使得原本活性极低的3d过渡金属如Cr、Mn、Co、Ni等,表现出良好的催化活性,甚至优于部分Ru基催化剂。进一步研究则发现,氢化物与过渡金属可在其界面处生成配位氢化物物种,而该物种可能是氮气活化转化的活性位点[64]。通过合成具有明确结构的配位氢化物Li4RuH6和Ba2RuH6,作者证实了配位氢化物可以作为一类新型的催化剂用于合成氨反应[65]。
在合成氨催化过程中,氮气和氢气共进料,由于固体催化剂表面对氮物种和氢物种吸附强度不同,不可避免地会产生氮气和氢气的竞争吸附问题。这一问题在金属钌表面上尤为突出。比如在传统Ru/MgO催化剂上,H2的级数为负值(-0.7),表明H2在Ru表面优先吸附解离,从而阻碍了氮气的吸附活化[66]。通过调变催化剂载体或添加助剂,可在一定程度上改变反应物的竞争吸附情况。另一种策略是采用化学链途径,即采取氮气和氢气分步进料的模式。化学链这一概念早在19世纪就曾被提出,如在加热的氮化钛上交替通入N2和H2实现了氨的生成[67]。Chen等[68]近期提出了一种以碱(土)金属亚氨基化合物为载氮体的低温化学链合成氨技术,即碱(土)金属的氢化物(如LiH,BaH2)首先通过“固定”N2生成相应的亚氨基化合物(如Li2NH、BaNH),随后将反应气氛切换为氢气使得亚氨基化合物加氢释放出NH3。值得一提的是,对于某些体系而言,其化学链产氨速率显著高于催化速率。比如,在相同反应条件下,BaH2/BaNH的化学链产氨速率是其催化速率的20倍[69];在过渡金属催化剂的协助下,BaH2/BaNH体系在250℃反应条件下的化学链产氨速率较高活性Cs-Ru/MgO的催化产氨速率高约1个数量级[68]。化学链过程与催化过程的另一显著区别是化学链过程是非平衡态过程,材料的相结构随着时间与反应条件的改变而发生变化。如在固氮过程中,碱(土)金属氢化物可形成相应的碱(土)金属亚氨基化合物。而在催化过程中,催化剂的相结构在一定时间尺度内可以保持不变。
在过去的一个多世纪里,人们在理解合成氨多相催化过程方面积累了大量的认识,在工业催化剂的设计和开发过程中已发挥重要的指导作用。然而令人感到遗憾的是,至今仍未实现“低温低压高效合成氨”的目标。借助电、光、等离子体等外场作用可能是实现温和条件下N2到NH3的高效转化的一种有效途径,更符合绿色合成氨的发展趋势。在外场驱动下,固体表面与N2分子间的电子传递及能量转换机制会发生变化,这可能有助于构建氮气活化转化新模式,以实施低温、低压、高效合成氨。此外,面对这一经典研究课题,我们需要新的思想。借助于凝聚态化学这一新兴学科的视角以及研究方法,对合成氨反应过程的多层次凝聚态结构、性质和功能开展系统研究,或将有助于我们更加深入地理解这一科学难题,形成新的催化剂研发理念。
4 氮气/氧气多能耦合转化中的凝聚态化学
氮氧化合物(NOx,如NO、NO2等),作为空气主分(N2和O2)的衍生物,是化工行业非常重要的原料。NOx的下游产品,如硝酸(HNO3)、三硝基甲苯(TNT)等,对于化肥和军事等领域至关重要,工业需求量巨大[70,71]。目前,合成NOx和其下游产品需要先通过工业合成氨,再经由NH3氧化得到NOx。该过程消耗的纯H2通常由化石能源制氢(包括水煤气变换、天然气重整等)获得,因此整个流程的能耗极高,且伴随着巨大的CO2排放[72,73]。如果能以空气为原料直接高效合成NOx,甚至直接合成含有C-N-O的高附加值有机化合物,将具有十分重要的意义。传统热化学的手段直接转化N2和O2因受限于N2分子弱极性以及高N≡N键能,十分困难。从凝聚态化学角度出发,探索不同的能量场(如光、电、声)或者多能耦合场与凝聚态物质(反应分子、催化剂)的相互作用模式与机制,对实现N2与O2的直接活化与转化制NOx具有重要指导意义[71]。在不同能量场输入过程中,化学反应往往会呈现出与传统热化学不同的形式,如不同能量场输入条件下,分子间反应路径不同,凝聚态材料在不同能量场下展现出的物化性质以及催化特性也不同。通过凝聚态化学的角度研究这类问题,可以更好地加深研究者对这些过程的理解,为更合理地设计空气的直接高效转化提供理论基础,同时也可以将凝聚态化学的广度扩展到物质与能量场的相互作用。本章将结合不同能量场下空气转化的研究工作,对这一类涉及物质与能量场相互作用的问题进行讨论,为其今后的发展方向提供一些借鉴。
4.1 低温等离子体过程
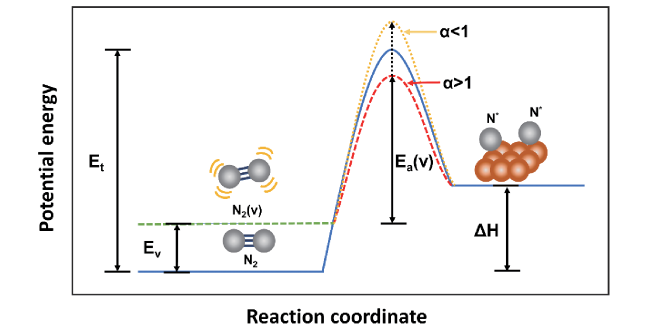
非热等离子体(Non-thermal plasmas, NTPs)由于其独特的非平衡特性,可作为温和条件下高效空气转化的可行方法[74]。NTPs由气氛分子、高能电子、激发态分子(如振动激发态和电子激发态等)和离子组成,其中高能电子的温度可以超过几千K,而宏观气氛分子温度可以保持在室温或者略高于室温[75]。因此,温和条件下热化学无法驱动的反应通常可以在NTPs条件下发生,并可借助NTPs与凝聚态结构电子态的耦合,实现高效的等离子体-催化反应过程。由于N2分子具有单一的振动方式、广延的振动能级分布和相对较大的振动能级间距(约0.3 eV),使得通过振动激发的方式促进N2的活化和解离成为可能[75]。一方面,振动激发态N2分子振动量子数的提升可以使N2分子通过能量不断累积的方式在气相中直接解离。另一方面,如图7 所示,振动激发态N2相较于基态N2可在一些催化剂表面上更容易地发生解离反应[76]。
e-+N2→e-+N2(v)
e-+O2→e-+2O·
N2(v)+O·→NO+N(v)·
N(v)·+O2→NO+O·
而在催化剂表面主要经历振动激发态N2的吸附解离,并且与O·自由基结合生成NOx[86]:
N2(v) $\mathop{ }_{\rightleftarrows}^{surface}$2N*
O·+N*$\stackrel{surface}{\longrightarrow}$NO*$\stackrel{desorption}{\longrightarrow}$NO
NTPs与催化耦合转化空气的过程十分复杂,涉及众多物理化学过程和催化剂-吸附质凝聚态相互作用,而这与NTPs中“态”的非平衡、多层次结构特性直接相关。同时,由于NTPs通过电磁场激发,这些凝聚态结构在电磁场中的特性也会对化学反应产生有重要影响。首先,NTPs中的粒子能量处于非平衡状态(电子能量>分子激发态能量>分子平动能),粒子能量的多层次结构使得粒子间的能量可以实现交换和积累,驱动N2分子的直接活化。此外,固体催化材料的催化特性在等离子体氛围下与热化学反应条件下往往不同。因此,在等离子体催化耦合中的催化剂-吸附质凝聚态相互作用,极大地影响着催化反应过程。如,研究者们已经证明了等离子体催化合成氨火山型曲线拐点的出现会滞后于热催化过程,合成氨速率更快且可以突破热催化条件下的热力学平衡限制[76,87]。这是由于振动激发态N2吸附在催化剂表面形成的凝聚态结构相较于相同平动能下吸附的N2有着更高的解离反应效率,从而使得合成氨的决速步从N2分子的活化解离转变为加氢和产物的脱附[87]。另外,凝聚态的存在也会影响等离子体的形成,如催化剂的形貌、介电性质等会影响凝聚态物质在高频电场下的极化,进而影响局部等离子放电。凝聚态结构由于局部阻抗不同会产生不同的局部热效应等,对于N2与O2反应的进行都有一定影响。
4.2 电化学催化过程
电化学催化反应包括反应物溶解、传质、吸附、活化、反应和解吸步骤,反应涉及气、液、固三相凝聚态结构的复杂变化。设计电化学反应,首先需要充分考虑电解液的性质、电极电位和电极材料的催化活性等因素。由于N2分子自身非常惰性,溶解度低,且在催化剂表面吸附强度弱,使得N2的电化学氧化十分困难[88]。对于电极反应,根据能斯特方程,析氧反应(OER)和氮氧化反应(NOR)的标准电位在常温常压下相对于标准氢电极(vs SHE)非常接近,分别为+1.23 V和+1.24 V,因而在以水为电解液的NOR体系中,OER竞争难以避免[89,90]。因此,调控溶液pH值和电极电位对于控制OER竞争反应至关重要。基于298 K和1 bar条件下在纯水或者1 mol·L-1溶质水溶液中建立的N2-H2O电位-pH图(Pourbaix diagram)[91],在溶液pH值高于1.3时,特别是在中性和碱性条件下,10电子转移的NOR过程(7)比4电子转移的OER过程(8)更有利[89,90]。
N2(g)+6H2O(lig) ⇌ +12 +10e-
O2(g)+4H++4e-⇌2H2O
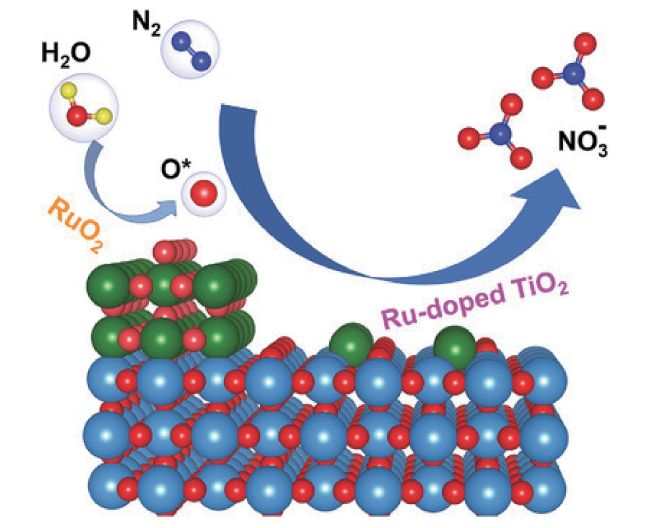
NOR不仅需要催化材料具有足够的活性,同时还需准确地平衡N2和H2O/OH-的竞争性吸附、活化、解离以及产物的解吸过程。因此NOR催化材料的设计相比于OER、氧还原反应(ORR)和析氢反应(HER)来说要求更高。理论上,NOR的反应速率取决于N2分子在催化剂表面活性位点上的解离能垒,同时也依赖于活性位点的数量。对于N2还原成NH3(NRR)过程,理论和实验都验证了N2解离是催化剂表面上的决速步[92⇓~94]。NOR过程与此类似,这需要催化剂具有高的N2分子吸附解离活性,同时考虑到电化学环境的氧化刻蚀效应,需要催化剂兼具高稳定性。贵金属因其未完全填充的d轨道、易吸附反应物和适中的吸附强度,是电化学催化领域的优异催化剂。早期,Zhang等[95]利用Pt电极在2.19 V vs RHE电位条件下成功实现了N2氧化到HNO3,然而其法拉第效率仅为约1.23%,单纯使用贵金属难以有效解决NOR过程的效率和选择性问题。Yan等[96]通过在TiO2上掺杂Ru,成功构筑了两种含Ru的活性中心(图8 )。其中掺杂在TiO2晶格内的Ruδ+可以有效活化N2并将其氧化为中间体NO*,而TiO2上的RuO2物种是电催化OER的活性中心,可以促进中间体NO*进一步氧化成HNO3。通过平衡两种活性位点的相对数量,NOR的法拉第效率可以达到26.1%。其他如掺杂型双功能催化剂[97]、尖晶石类氧化物催化剂[98]、负载型二维材料催化剂[99],都可以实现比纯贵金属催化剂更高的法拉第效率。
目前,电催化NOR过程仍处于基础研究阶段。从凝聚态化学角度,研究催化剂表界面与反应物、活性中间体和产物的凝聚态相互作用机制非常关键。在阳极,大多催化剂表面更倾向于吸附OH-,即使N2分子可以在一些催化剂表面被优先地吸附,其后续通常也难以被有效活化,并可能因OH-的竞争吸附而解吸,最后发生OER过程。但是,保持适当的OER活性却可以加速NOR过程,通过合理地设计催化剂从而分离OER与NOR活性中心,构筑分别用于OER与NOR的两种相邻活性位点并平衡其数量,是可行途径之一。同时也需要注意,并非所有活性氧物种都能与吸附的N2结合形成NO,大多数活性氧物种会与其他氧物种(如OH-等)结合形成O2。所以理清不同电位下N2与OH-等物种的优先吸附模式以及凝聚态相互作用至关重要。这也要求研究者们在设计催化剂的过程中充分考虑催化剂复杂性和多样性的凝聚态结构所带来的在电化学条件下的各种效应(如电子效应、尺寸效应、应变效应、配体效应、边界效应、扩散效应等),而不再是单纯考虑活性位点。
4.3 超声转化过程
超声波是一种频率高于可听范围(约20 kHz以上)的声波。当超声波在液体介质中传播时,二者之间的相互作用会产生一系列物理化学效应,即超声效应,包括机械效应、热效应、空化效应和化学效应。其中,超声空化可以在局部产生极高的瞬时温度、压力以及声致发光[100,101]。此外,空化过程中产生的自由基可以引发化学反应,即所谓的超声化学效应。这一涉及复杂凝聚态结构变化的过程可以描述为在充满液体介质蒸气和气体分子的空化气泡内部,分子被高温高压环境活化并进一步解离[102⇓⇓~105];在气泡内部或两相界面,产生的自由基可以与反应物分子结合形成产物。因此,在超声场作用下,在空气饱和的水介质中N2可以被直接氧化为亚硝酸和硝酸。
早在1936年,Gohr等[106]就发现在频率为540 kHz的超声场作用下,H2O2、HNO2和HNO3可以在空气溶解饱和的水溶液中生成。后续的相关工作探索了不同条件(频率、pH值等)对于超声空气转化合成HNO2和HNO3的影响,以及相关的反应机理[107⇓⇓⇓⇓⇓~113],其中对于N2的活化方式有一定争论。David等[110]认为在超声空化气泡高温高压微环境中产生的·OH和·O都可以氧化N2分子,而也有一些研究认为·OH并不能直接氧化 [108]。无论如何,研究人员普遍认为超声场作用下产生的空化气泡内部的高温高压(约5000 K, 约500 atm)环境足以使O2和H2O等分子产生自由基,从而驱动N2转化反应。这就使得超声场下的溶液也呈现出类似于低温等离子体的特性,即整体溶液温度可能略高于室温,但是局部溶液微环境处于高温高压状态,使得反应物凝聚态结构发生变化。同时溶液的性质和状态,如溶剂特性、pH、阴阳离子类型、是否添加固相催化剂等,也可以影响空化气泡的成核和破碎、自由基的种类和数量,进而直接影响N2氧化的反应过程。
4.4 光催化转化过程
以太阳光为能源的光催化转化N2和O2提供了一条非常理想的替代传统基于化石能源进行空气转化的路径。作为典型的凝聚态反应过程,光催化N2氧化过程可分为以下步骤:首先,通过光催化材料吸收光将电子从价带(VB)激发到导带(CB),在VB中留下空穴(h+);然后,光生空穴h+以H2O为氧源将N2氧化为NO,O2通过光激发电子被还原成H2O;NO可以进一步被O2和H2O氧化成硝酸或者硝酸盐。早期的光催化固氮研究主要聚焦在光催化转化N2合成NH3过程。而在2013年,Yu等[116,117]首次报道了在紫外线或太阳光(UV/Vis)照射下,空气可以在纳米TiO2表面上直接形成硝酸盐,并且FTIR差分光谱和理论计算结果显示硝酸盐的生成是一个逐步氧化的过程,其中NO*是中间产物。随后,Zhang等[118]使用Z型异质结TiO2/WO3纳米棒作为光催化剂,在光热协同作用下,也实现了N2氧化生成NO(产率0.16 mmol·g-1·h-1),在365 nm下的量子效率为0.31%,这也证明了NO是光催化N2氧化的中间产物。
光催化N2氧化需要在半导体的光生空穴h+上进行,同时反应还具有较高的能垒。因此,在设计相关催化剂的过程中,除了要考虑材料本身的电子性质,还需要考虑催化剂的整体结构设计,如通过掺杂、构筑缺陷、形貌修饰等手段,来促进N2分子与催化剂的光生空穴h+的快速接触并结合,形成关键的吸附凝聚态结构。Xie等[119]设计合成了富含坑洞结构的WO3纳米片,这种富含坑洞的结构不仅提高了N2与催化剂的接触面积,更重要的是其相对于单空位WO3具有更多的悬键,可以有效活化N2分子,同时更容易激发出高能电子,可以有效为后续活化的N2分子的氧化提供能量,即通过修饰WO3的形貌,可以克服N2光催化氧化的两个难点:N2分子的活化以及反应涉及的高能垒。这种富含坑洞结构的WO3纳米片可以在室温条件和380 nm的UV/Vis辐射下,以1.92 mg·g-1·h-1的速率生成硝酸盐,表观量子效率(AQE)为0.11%,优于无坑洞纳米片和块状WO3。Zhang等[120]发现将Cu单原子分散在TiO2晶格中,可以有效提升光生载流子的分离和转移效率,促进N2在Cu位点上的吸附与活化,以及后续氧化。在365 nm的光辐射下,可以达到0.93 μmol·h-1的硝酸盐生成速率,同时光谱表征验证了单原子Cu表面吸附的NOx(包括NO2+、NO+或NOδ+)中间物种(图9 )。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
目前,在温和条件下通过光催化转化空气制备硝酸盐依旧受限于产物生成速率和表观量子效率。在设计催化剂的过程中不仅需要充分考虑催化材料本征凝聚态性能,还需要充分考虑如何构筑催化剂的结构,因为元素相同的材料在微观结构不同时可能展现出不同的光催化特性;也可以通过掺杂手段修饰凝聚态电子结构,提升光生载流子的分离和转移效率,增加光催化活性位点数目等,促进凝聚态活性中间体的形成及其后续氧化过程中的结构变化。此外,已有研究证明光子能量与热能也存在耦合与协同作用,光热耦合催化也是转化空气的有效途径之一[121]。
5 结论与展望
一个多世纪以来,人类在固氮领域取得了巨大的成就,客观上也丰富了学界对凝聚态化学的认识。但是,这个领域依然存在许多亟待回答的问题。例如,在含有添加剂、盐以及配位溶剂的真实体系中,氮气与活性金属簇合物形成的凝聚态结构究竟是什么?从氮气转变为含氮物质是一个多步过程,随着氮气衍生化反应的进行,关键中间体的凝聚态结构是如何变化的?在凝聚态化学研究的第三层次,尤其是固态宏观结构层次[3],我们是否能够利用自组装原理发展新型的合成氨催化剂,使得反应物种的吸附能可以实现动态优化调整,从而规避催化中广泛存在的能量限制关系的限制,实现温和条件下的氮气高效转化呢?反过来,凝聚态化学的发展也将给固氮研究带来新的思想认识。通过凝聚态化学的角度,可否更合理地设计通过多能耦合的方式实现空气的直接高效转化,将凝聚态化学的广度扩展到物质与能量场的相互作用?
本文并不是一个完整的总结,而是作者基于自身理解,对于固氮化学领域中存在的一些凝聚态化学现象的粗浅分析。我们相信,随着表征技术的提升以及理论计算能力的加强,未来凝聚态化学的概念在固氮领域会得到越来越多的关注,并最终促进某些关键基础科学问题的解决,为人类开发氮气利用新途径提供思路。