




1 引言
高级氧化技术 (AOPs)[5,6]是目前处理工业废水最有效的手段之一,其通过光照、超声、电、磁等物理化学过程产生大量具有强氧化性的自由基,如羟基自由基 (·OH)[7]、超氧自由基 (· )[8]、硫酸根自由基 (· )[9]等,实现环境中有机污染物高效降解,具有无选择性、反应周期较短等优势。其中,基于半导体材料的光催化技术,具有反应条件温和、无二次污染以及利用太阳光等特点[10],在挥发性有机污染物 (volatile organic compounds, VOCs)[11,12]、持久性有机污染物 (persistent organic pollutants, POPs)[13]等污染物的处理中展现出独特的优势。然而,现有的光催化技术往往只关注有机污染物的矿化效率,忽视了有机物降解过程中的化学能利用。从能量角度看,污水中有机组分含有丰富的化学能,例如苯酚降解可释放3050.6 kJ·mol-1的能量。如果能将污染物降解过程中的化学能加以转化利用,则可以实现污染物降解和能量回收的双重效益。
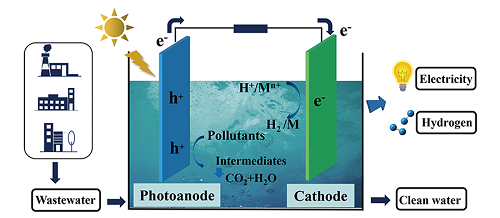
为了解决有机污染物的资源化利用问题,研究人员提出了光催化燃料电池 (photocatalytic fuel cell, PFC)[14⇓~16]的处理技术。PFC装置在保留传统微生物燃料电池[17,18]特性的基础上,通过利用半导体材料作为电极,结合光催化高级氧化技术的优点,使得体系中自由基生成数量以及电子传递效率大大提高,在污染物处理和能源回收等方面表现出了巨大的应用潜力。PFC装置中阳极材料通常为n型半导体,如TiO2[19⇓~21]、Fe2O3[22]、WO3[23,24]、BiVO4[25]等纳米薄膜电极,阴极则为Cu2O[16,26,27]等p型半导体或Pt、碳棒等电催化剂[28]。在光照条件下,阳极半导体材料被光激发后能够迅速产生光生电子e-、空穴h+,阳极处h+(或其反应生成的活性氧物种)发挥氧化作用高效降解污染物,e-自发移动至阴极发生还原作用。光能和化学反应驱动的闭合电路电流循环过程产生了电能,实现了污染物化学能回收。本文从PFC的原理和应用入手,综述了PFC在环境污染物无害化处理的研究进展,并详细介绍了提高PFC的污染控制性能和产电效率的调控手段。
2 PFC的结构与工作原理
通常,PFC装置由两电极(阴极、阳极)、外部回路、电解液、燃料(有机物)以及离子交换膜等部件组成(图1 ),其中,电极材料通常包括光响应半导体催化材料和具有良好导电性能的光惰性电催化材料两大类。根据阴极电极材料的差异,PFC可以被分为单光电极PFC和双光电极PFC两类。整个装置中,半导体电极材料发挥着核心作用。半导体的能带结构中包含导带 (conduction band, CB) 和价带 (valence band, VB) 结构,n型半导体费米能级靠近其CB,而p型半导体的费米能级则在VB附近,半导体材料的费米能级决定了电极所处的电势位置。半导体未被激发时,e-分布于价带内,当半导体材料吸收了能量大于禁带宽度的光子后,价带中的e-被激发进入导带,价带中留下带正电荷的h+(式1)。h+具有较强的氧化性,能够与水溶液中H2O或OH-反应生成具有强氧化性的·OH(式2、3),在阳极处发挥氧化作用;由于阴极电势高于阳极电势,电子能够自发地由阳极材料移动至阴极表面,电子可以与阴极表面的O2反应生成· (式4)、H2O2等氧化性物质,也可以还原氢离子 (H+) 或者重金属离子 (Mn+)(式5、6),起还原作用。
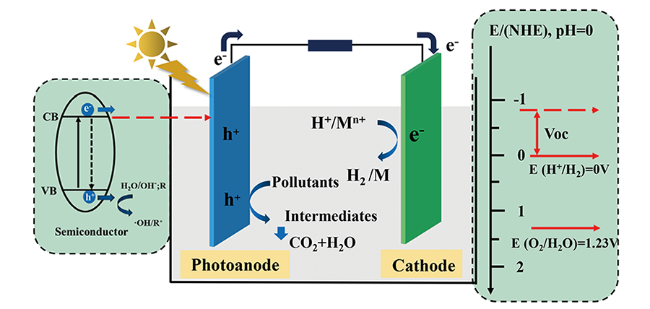
半导体的能带结构对PFC的性能影响较大,原因如下:(1) 禁带宽度决定着半导体对光能的利用能力,带隙较宽的半导体如TiO2 (3.2 eV),只有在能量较高的紫外光照射下才能激发,其对太阳光的利用率小于5%[29];(2) VB的位置决定了阳极h+的氧化能力,只有当VB电势高于OH-/·OH的氧化电位2.8 V (vs NHE, pH=0) 时,·OH才能够生成,例如Wang等[30]合成的BiVO4的VB电势为2.12 V,高于析氧反应O2/H2O的氧化电位1.23 V (vs NHE, pH=0),此时体系可以发生水解析氧反应,但低于OH-/·OH的氧化电位,光催化过程中无法生成·OH。因此,设计具有合适能带结构以及高载流子分离效率的半导体材料,是提高PFC性能的关键因素之一。
衡量PFC装置性能的指标除了污染物降解效率以外,还有开路电压 (open-circuit Voltage, Voc)、短路光电流密度 (short circuit photocurrent density, Jsc)、最大功率密度 (maximum power density, JVmax)以及填充系数 (fill factor, FF) 等关键产电效率参数。理想条件下,Voc等于PFC阳极和阴极之间的电势差,选择合适的半导体材料以及设计合理的阴极还原反应有利于提高Voc。Jsc可以间接反映体系中的由阳极传递至阴极的电子数量,阳极空穴的有效消耗以及阴极电子的迅速转化,可以有效提高体系的Jsc以及JVmax[31]。填充系数FF指的是电池的实际最大功率密度与理论最大功率密度之比(式7),表明了体系中各项损耗对PFC性能的影响。提高体系的电子传递以及传质效果能够有效提高体系的FF数值。
3 PFC的分类
3.1 单光电极PFC
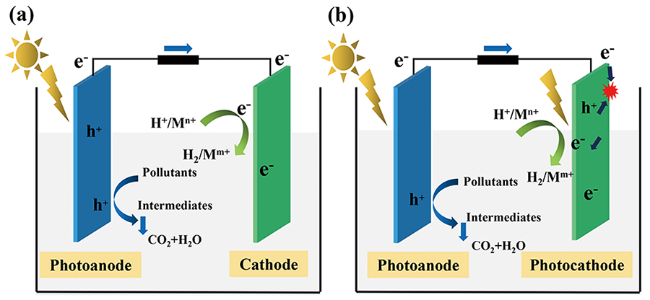
单光电极PFC系统的阳极通常为n型半导体,阴极则为光惰性电催化材料(图2 a)。阳极的电势取决于材料的费米能级;阴极的电势取决于阴极发生的还原反应的氧化还原电位。阴极发生的还原反应与阳极处的有机物种类无关,通常只受到体系pH值与O2含量的影响(式8~11)。
无氧环境:
有氧环境:
Kaneko等[14]首次构建了工作电极为TiO2的PFC装置,该体系中阳极可以有效处理多种生物质材料(如多聚糖、蛋白质、纤维素等),类生物质材料甚至聚合物,阳极氧化过程中产生的质子转移至空气呼吸阴极表面发生氧还原反应。该体系中阳极半导体可见光利用率低。后续研究多致力于改进半导体电极材料以及优化阴极还原反应。
半导体的能带位置 (CB) 决定了阳极的电极电势。常用的单光电极PFC系统中,光阳极半导体材料包括TiO2[19,21] (CB = - 0.2 V vs NHE)、ZnO[32,33] (CB = - 0.3 V vs NHE)、Fe2O3[22] (CB = + 0.5 V vs NHE)、CdS[34] (CB = - 0.5 V vs NHE)、ZnS、WO3[16,23,24] (CB = + 0.3 V vs NHE) 等。此外,阴极电极材料所处环境对PFC装置能否正常运行非常关键。如Fe2O以及WO3等半导体的CB远高于析氢反应的还原电位 (0 V vs NHE, pH=0),此时,不合适的电势差无法驱动电子自发移动。当阴极有氧气存在时,阴极反应的还原电位为1.23 V (vs NHE pH=0),此时,阴极电势高于阳极电势,PFC中电子可以在内部电势差的驱动下自发移动。
3.2 双光电极PFC
用半导体材料替代传统的惰性阴极材料,形成双光电极PFC,能够进一步提高体系的光利用率。双光电极PFC装置中,阳极通常为n型半导体,阴极为p型半导体。如图2 b,两电极材料均可以在光照条件下产生光生e-和h+。阳极的e-自发移动至阴极后,与阴极半导体材料产生的h+发生猝灭作用,阳极的h+与阴极的e-分别在两电极材料表面发挥氧化作用以及还原作用。
Cu2O是PFC装置[16,26,27]中最为常见的阴极半导体材料之一,其带隙宽度为2.0 eV,具有良好的可见光吸收能力,制备方法简单且成本低廉。Zhou等[16]构建了阴极为Cu2O/Cu的双光电极PFC体系,在可见光照射下,体系的开路电压可以达到0.21 V。相比之下,单光电极存在时,体系的开路电压几乎为0。为了提高Cu2O材料的光生载流子分离率以及提高材料的稳定性,研究人员开发了CuS/Cu2O[35,36]、Cu2O/CuO[37,38]、Cu2O/NiOX[39]等Cu2O复合材料,应用于PFC光阴极。另外,MoS2[40,41]、BiOBr[42]以及Pt/PVC[43,44]等也被大量用作PFC光阴极材料。
构建双光电极PFC系统能提高体系的污染物去除效果和产电效率。Zhao等[26]利用C/Cu2O/Cu替代Pt作为阴极,在模拟太阳光的照射下,PFC体系的性能明显提高,对苯酚的降解率从6.4%提高到84.2%;Yu等[45]构建了BiVO4/TiO2纳米管阵列阳极、Cu2O/TiO2纳米管阵列阴极的双光电极PFC,双光电极PFC中污染物降解速率常数分别是BiVO4/TiO2纳米管阵列阳极-Pt阴极体系、Pt阳极-Cu2O/TiO2纳米管阵列阴极体系的1.42倍和3.66倍,最大电流密度分别提高了1.68倍以及103.8倍。双光电极的引入提高PFC的性能,主要原因如下:(1) 提高了体系对于光照的利用率,提高了体系中载流子的生成量;(2) 阳极e-迁移到阴极与h+复合淬灭,有效提高了阳极h+和阴极e-的分离效率和寿命,有利于发挥氧化和还原作用;(3) 相较于单光电级体系,减少了阳极产生的电子传递至阴极的电子能量的损耗;(4) 光阴极表面电子所处能级更高,有利于电子发挥还原作用。
4 PFC体系的性能优化
PFC装置仅需太阳光的能量输入,即可实现阳极氧化、阴极还原以及产电的多重效益。如何有效地提高PFC装置的污染物降解以及产电性能,是后续研究关注的重点。本文从半导体电极材料的优化、电催化阴极材料优化、反应器装置以及操作条件等四个方面较为详细地概括了提高PFC性能的各项方法以及策略。
4.1 PFC半导体电极材料的优化
光响应半导体电极[46]是PFC装置的核心组件。光生电子和空穴的数目以及活性决定了PFC装置对污染物的降解效率以及能量的产出效率,因此,提高半导体材料的光生电子-空穴对生成速率以及减少电子空穴对的复合成为优化PFC电极材料的关键点。常见的半导体材料改性途径包括:形貌优化、材料表面调控策略(贵金属沉积、半导体复合、晶面调控、元素掺杂和缺陷调控)、材料-电解液界面反应工程等,这些手段同样也适用于PFC光电极的优化工程。
目前,调控光催化剂的形貌是半导体材料常用的改性方法,通过构造纳米线[47]、纳米管[48]、纳米棒[49]以及纳米球等二维或者三维形貌的功能纳米材料,可以有效提高材料的光催化性能。形貌调控的优势包括:(1) 提高了材料的比表面积,为反应提供了更多活性位点。Li等[50]合成了花状微孔空心球状的BiOBr,其比表面积可以达到31.50 m2·g-1,相较于传统方法合成的BiOBr,其光催化降解污染物效率提高了18%。(2) 提高光折射,促进光吸收。Wang等[51]通过调控合成过程中S元素和I元素浓度比,得到了纳米片、纳米棒和纳米花状等不同形貌的Bi2S3/BiOI-TiO2阳极材料,并应用于PFC体系,结果表明,花状复合半导体材料具有更高的可见光吸收率,应用于PFC装置,其对于Cr(VI)去除率是片状材料以及棒状材料的1.25倍和1.82倍,对有机底物RhB的降解反应速率常数提高了1.38倍以及1.87倍。(3) 促进电子的定向传递,降低了材料载流子复合,如Qi等[52]合成了不同长度的TiO2纳米管阵列材料,相比于颗粒状材料,纳米管阵列形貌大大降低了材料的载流子复合,TiO2纳米管阵列的载流子寿命提高了3.37倍,该PFC体系得到的最大Jsc高达1.572mA·cm-2,最大FF达到0.84。
另一种常用的半导体改性方法是对催化剂进行表面改性,包括贵金属沉积、半导体复合、晶面调控、制造缺陷等。贵金属粒子(如Au[53,54]、Ag[55,56]以及Pt[57]等),具有局域表面等离共振效应,能够提高材料的光响应范围,有效促进半导体载流子分离。Dector等[58]通过将Au纳米颗粒沉积在ZnO表面作为PFC阳极材料,紫外可见吸收光谱表明Au沉积后大大提高了半导体材料在415~710 nm范围内可见光吸收能力,这主要源自于Au粒子的局域表面等离共振效应。表面沉积另一种窄带隙的半导体材料形成异质结结构也是常用的半导体表面改性手段之一。异质结主要可分为p-n异质结[59]和Z型异质结[60],异质结的形成可以促进光生载流子有效分离。例如Xie等[61]构建了阳极为WO3/BiVO4异质结的PFC装置,BiVO4的带隙宽度为2.4 eV,相较于WO3 (3.2 eV),异质结的存在极大地提高了阳极对可见光的吸收能力,且二者合适的带隙结构,能促进电子转移到WO3的CB上,空穴转移到BiVO4的VB上。该研究中,在模拟太阳光以及1.2 V (vs Ag/AgCl) 的外置偏压下,WO3/BiVO4材料光电流可以达到5.04mA·cm-2,为纯WO3的2.84倍,紫外可见吸收光谱也表明,形成异质结后,材料对400~500 nm波长范围内光的吸收能力得到了提升。类似的异质结半导体阳极材料近年来也逐渐被运用到PFC体系之中,如WO3/g-C3N4[62]、TiO2/WO3[63]、TiO2/BiVO4[64]、MnO3/ZnO[65]等。半导体表面原子的排列以及配位能够很大程度影响反应物分子的吸附、和反应物分子之间的电子转移以及产物的脱附过程。通过有目的地调控半导体表面的晶面排布方式,也可以有效提高材料的反应性能以及选择性[66]。Jia等[67]研究了 (010) 晶面BiOCl在PFC系统中更易发生染料光敏化的原因,发现 (010) 晶面的特殊结构有利于电子的转移,并且,其拥有更大的比表面积和活性位点,有利于有机物的吸附与降解。这表明合成高 (010) 晶面含量的BiOCl能够有效提高PFC系统性能。此外,合理构建晶体结构缺陷也可以有效提高材料的光催化性能,一方面,构建体缺陷可以调控半导体的带隙结构从而提高材料的光吸收范围;另一方面,材料表面的缺陷可以成为催化反应的高效活性位点[68]。构建合适的缺陷,例如氧缺陷[69]、Bi缺陷[70]等,也是有效提高光催化半导体材料活性的重要手段之一。
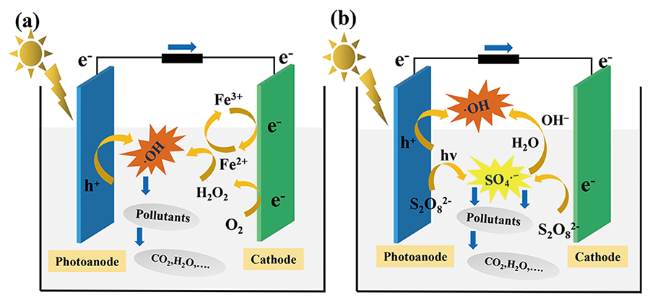
除了以上对于半导体材料本身的改性和优化外,光催化半导体的材料-电解液界面反应工程也同样作为提高PFC体系性能的一种重要途径。光催化反应装置中,基于自由基的氧化还原反应只能发生在电极材料和溶液的固-液界面处,扩散距离非常有限,这也限制了PFC装置对有机污染物的降解以及产能效果。常见的材料-电解液界面反应工程策略包括:芬顿反应耦合、硫酸根自由基耦合等。通过向PFC体系中添加亚铁离子Fe2+[71]或者过硫酸盐 (persulfate, PS)[72]可以有效提高体系中的自由基种类以及活性。芬顿反应耦合PFC体系中,阴极表面氧还原反应产生的H2O2与Fe2+反应生成·OH(式12、13)(图3 a)。Xu等[73]在WO3/W阳极、Fe@Fe2O3/碳毡阴极构成的PFC装置中引入Fe2+,通过芬顿体系的耦合,增强了阳极电子空穴分离效果和体系中的自由基反应。相较于单一的光催化和电芬顿催化反应,芬顿反应耦合的PFC体系对于污染物降解速率分别是前者的2.07倍以及1.71倍。Fe2+通常在酸性条件下活性较强,而在较高pH条件下,Fe3+难以转化为Fe2+。为了改进中性甚至碱性环境下Fe2+转化率不高的问题,Zhou等[74]在原有的PFC体系中引入WS2共催化剂,WS2分子中W4+具有还原性,能够协助Fe3+的转化,有效促进了体系中Fe2+/Fe3+的循环。而通过在PFC系统中加入PS,S2 与电子反应,一方面可以快速消耗催化剂表面的电子,促进了电子和空穴的分离;另一方面反应生成· ,· 在溶液中自由移动,可以引发一系列的自由基反应,从而提升污染物的降解效果(图3 b)。Li等[75]在阳极为TiO2纳米棒、阴极为Pt的PFC装置中,引入了PS,以促进抗生素诺氟沙星的降解和体系产能发电效果,PS和PFC体系形成了良好的协同效应,耦合PS/PFC体系的发电量约是单独PFC体系的两倍。
4.2 PFC光惰性阴极材料的优化
Pt化学性质非常稳定,且导电性能优异,是传统PFC装置中最常见的阴极材料。Pt作为一种贵金属,其昂贵的价格限制了其大规模推广使用。目前,商业化的Pt/C催化剂活性较好、选择性优良、导电性能良好和比表面积较大,是一类较好的Pt电极替代品,广泛应用于诸多PFC体系中[32,79]。此外,制备PtM(M代指金属)合金[80,81],也是重要的设计Pt电极替代品手段之一。一些常见的有机导电聚合物,如聚吡咯 (PPy)[28]、聚吲哚、聚苯胺等,因为具有高导电性、较快的载流子迁移率、高的吸收系数以及较强的环境稳定性,成为了Pt电极的优良替代品。Jia等[82]开发了PPy负载的Ni箔作为PFC阴极析氢材料,PPy膜显著降低了Ni箔的析氢过电位,Ni箔本身具有的高比表面积也为析氢反应提供了更多的活性位点。二者结合对于体系中电极对于质子的捕获以及转化具有较好的协同效果,重复性实验(13次)也表明该材料具有优异的稳定性。
通常PFC装置中有机污染物的氧化主要依赖于阳极的氧化作用,阴极表面发生的二电子ORR产生的H2O2没有得到充分利用。使用改性的碳基材料可以有效提高阴极二电子ORR反应的效率。Zheng等[83]利用 (NH4)3PO4 处理竹炭 (BC) 得到新型功能性阴极材料,丰富的N、P以及含氧官能团使得材料具有优异的产H2O2性能,体系中产生的H2O2与Fe2+发生芬顿反应转化为·OH等自由基,有效提高污染物的降解性能。Zhou等[84]使用H2SO4活化的碳砧 (CF) 作为PFC装置中的阴极电极,H2SO4活化使其表面产生丰富的含氧官能团,这些含氧官能团作为ORR反应的活性位点,生成H2O2并转化为·OH以及· 等活性物种,提高了PFC装置的性能。该装置相比铂碳材料作为阴极的PFC体系,JVmax提升了1.69倍,污染物的降解效率提高了5.15倍。由此可见,碳基材料可以有效提高阴极ORR反应,在提高体系污染物降解效果上具有独特的优势,并且价格低廉且原料易得,应用前景优异。
4.3 PFC反应器结构的优化
PFC问世以来,诸多研究致力于研发高效的催化电极材料。然而,PFC反应器结构对于催化反应过程中体系的光利用率以及催化反应活性的影响作用同样不容忽视。除了传统的单室反应器外,研究较为广泛的还有“H”型反应器(图4 a)、转盘式反应器(图4 b)以及微流体反应器(图4 c,d)等,后两者可以有效提高体系传质效果、光吸收能力等,从而有效提高PFC的综合性能。
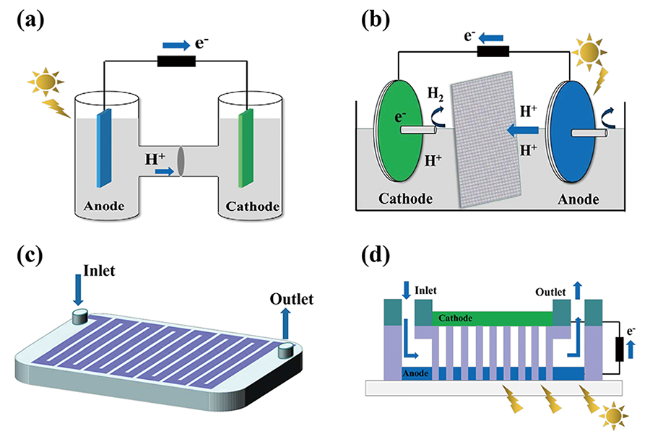
4.3.1 H型反应器
传统的H型PFC反应器结构较为简单(如图4 a),两电极分别处于被离子交换膜隔开的单独反应腔室中,可以通过曝气或者磁力搅拌增大体系传质效果。该反应器一般容量较大,且能够将燃料固定在阳极腔室内,避免了阳极中氧化反应和阴极的还原反应之间的互相干扰。阴极与阳极有效隔绝,可以促进阴极析氢或者重金属还原反应的发生。Kamat等[85]构建的PFC体系,使用自制的Nafion膜后,可以实现流动态操作条件下产氢,速率可达69 μL·h-1·cm-2。
H型反应器也有一个明显的缺点,即两电极之间的间距较大,从而导致体系的电阻也较大,电池的FF较小。Lianos等[86]在双腔室光催化燃料电池的研究中发现,当两电极的距离为17 cm时,每个腔室的容积可达约200 mL,以甘油作为燃料时,该电池的FF从0.61下降到0.28。为了验证是否为电池的内阻过大造成了功率的损失,他们又组建了一个两电极距离仅为6 mm,容积仅为数毫升的双腔室光催化燃料电池,且其他条件保持不变,燃料电池的FF仅从0.65下降到了0.59,极大提高了装置的产电效率。如何有效减小体系内阻,提高PFC装置性能是H型反应器需要重点关注的一大问题。
4.3.2 转盘式反应器
传统的单室以及双室反应器,由于电极材料浸没在溶液之中,溶液对光的吸收以及反射大大降低了体系中光的利用率,且需要额外增加曝气装置以提高体系的传质效果。针对上述问题,转盘式PFC反应器(图4 b)表现出以下优势:(1) 在转盘式反应器中,光电极被制作成圆形盘片状,盘片在光催化反应过程中,保持一定速度旋转,电极的下半部分浸没在溶液中,上半部分则被一层水膜包覆,旋转使得水膜得以持续更新,保证了底物得以有效利用;(2) 由于光照不需要通过原溶液,仅需要透过一层水膜就能被电极吸收,这一过程中光的辐照损失可以大大降低;(3) 盘片的转动可以带动水膜持续更新,这也大大增加了体系的传质效果。
转盘式反应器设计近年来被广泛应用于PFC体系之中。Jia等[87]对比了转盘式水膜PFC与传统浸没式PFC性能的差异,得出转盘式PFC的产氢能力是传统式的10倍,其对污染物的降解效果和发电能力也远远高于传统反应器。他们后续也进行了诸多转盘式反应器应用于PFC体系的研究[88,89]。Zheng等[90]利用转盘式光催化燃料电池处理气态甲醛,运行75min后甲醛的吸收率达到了94%,降解率达到了82.4%。通过对比,转盘式光催化燃料电池对甲醛的降解效果是非转盘式PFC的2.3倍,是传统气相处理系统效果的3.6倍。转盘式设计增强了体系对污染物的吸附效果和氧化速率,在不同湿度条件下,对甲醛均具有较高的处理效果。PFC体系中阴极和阳极均可以设计为转盘式结构。Li等[91]利用不同形貌的纳米聚吡咯阴极电极和TiO2纳米管阳极,组装成双转盘式PFC,该转盘式PFC对实际纺织废水表现出优异的处理效果,体系的Voc为0.44 V,Jsc为2.00mA·cm-2,JVmax为0.22mW·cm-2,COD的去除率约为74.1%。
4.3.3 微流体反应器
微流体反应器是利用精密加工技术制造的当量直径为微米到毫米尺度之间的流体通道反应器。该结构反应器具有比表面积大、扩散距离短、辐照均匀、反应周期短、易于调节控制反应参数等优点,能够大大提高反应器处理效率。此外,微流体反应器可以实现流动式污水处理,其单位装置处理污染物的容量虽然有限,但通过并联、串联多个反应器可以有效扩大装置的污水处理量。
鉴于微流体反应器的诸多优势,构建微流体PFC体系成为了近年来的研究热点。如图4 c,d所示,微流体PFC反应器中,阳极材料负载在反应器流体通道表面,有效提高材料与反应溶液之间的接触面积;物料以流动态通过整个反应器内部腔室或者微流体通道,由于微流体尺寸结构微小,物料能够快速扩散到电极表面;微流体内部物料流动呈较为理想的层流状,各部分具有较为均一的处理效果,可以通过控制流速较好地调控反应器的工作性能。
微流体PFC装置有效提高系统污染物处理效率和产能效果。Chen等[92]构建了一种无膜、空气自呼吸阴极的微流体PFC装置,以葡萄糖以及MB作为模拟污染物检验了该系统的可行性。TiO2/FTO阳极电极负载于微流体反应器底部内壁,光照透过FTO玻璃可以直接激发催化剂表面产生光生载流子,另一侧为自呼吸碳纸阴极。该微流体反应器的设计较好地解决了传统PFC装置中传质率低、体系内阻大、表面积小的缺点,其阴极处无需额外曝气,也简化了装置构造,更利于实际应用。在该系统中,发现提高污染物浓度以及光照强度可以有效提高PFC装置的最大功率密度。污染物浓度从0.05 mol·L-1提高到0.3 mol·L-1,PFC装置最大功率提高2.73倍;而光照强度从22mW·cm-2提高到112mW·cm-2,Jsc提高3.19倍。他们随后研发了基于CdS/ZnS-TiO2为光电阳极,CuO为光电阴极的双光电极微流体PFC体系[93],该装置中,阴极和阳极在空间上并列排布,使得两光电极均可以利用一侧分布的光源。提高甲醇浓度 (0.05~4mol·L) 可以有效提高体系的Jsc (3.11倍)以及JVmax(2.77倍)。值得注意的是,当流体流速从10 μL·min-1提高至200 μL·min-1,体系的Jsc以及JVmax分别从2.1mA·cm-2、0.51mW·cm-2提升至2.3mA·cm-2以及0.64mW·cm-2,当流速从200 μL·min-1提高至1200 μL·min-1,Jsc以及JVmax分别下降到2.1mA·cm-2以及0.51mW·cm-2。原因是流速会影响污染物的停留时间以及体系的传质效果,选择合适的流速实现二者平衡对微流体PFC的工作效率至关重要。
微流体反应器提高了PFC系统的光吸收效果以及传质效果,且可以实现处理流动相污染物。因此,微流体反应器在航海船舶的废水处理、航空航天废弃物处理以及医疗器械供电等特殊领域具有非常广阔的应用前景。近年来甚至报道了将微流体PFC系统应用于一些便携式医疗设备中供电的应用,例如,Dector等[58]研发了利用血糖以及空气中的氧气作为燃料以及氧化剂的微流体PFC,可见光条件下,装置的Jsc可达到0.63mA·cm-2,JVmax可达到0.13mW·cm-2。
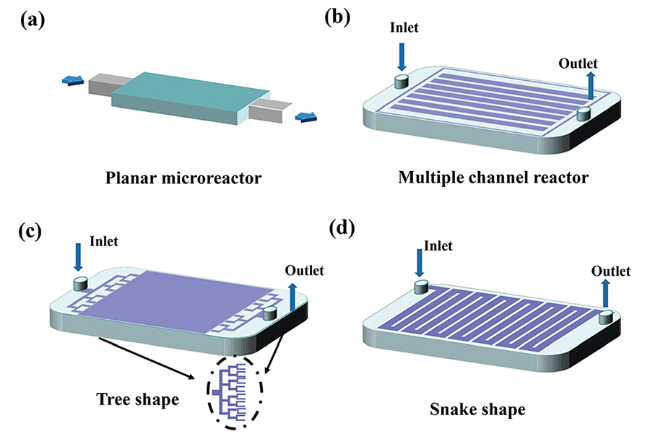
微流体反应器的流体通道类型对反应器的性能影响也较大[94]。常见的微流体反应器通道类型有“平板”型[95](图5 a)、“多通道”型(图5 b)、“树枝”型[96,97](图5 c)以及“蛇”型[98,99](图5 d)等。合理设计微流体反应器的流体通道可以有效提高反应器的比表面积,提高体系的传质效果以及降低流体流动过程中的传质阻力[100]。目前,为了探索微流体反应器流体通道对系统效率的影响规律,除了设计实验以外,流体力学模拟计算也被大量使用。COMSOL以及ANSYS FLUENT等计算流体学仿真软件广泛应用于催化剂膜层厚度[101]、流体流速等操作条件优化方面的研究,通过仿真模拟还可以得到体系中各组分浓度[102,103]、温度[104]、速度[105]以及压力[106]分布情况,为设计更加高效合理的微流体通道结构提供理论指导,如Luo等[107]基于ANSYS FLUENT三维CFD仿真数据,设计了一种Z型梯形流体结构,该通道设计结构具有优异的流体分布均匀性;Leung等[108]相关研究通过模拟计算以及实验验证表明优化的电流集电极设计可以有效地降低多孔电极流动MFC中的欧姆电阻,从而提高电池的性能。近些年来激光雕刻或者3D打印[109]等技术广泛被运用到微流体通道的设计中,这些技术能够较大提高通道设计精度。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
4.4 其他反应参数的影响效果
4.4.1 氧气
体系中的溶解氧 (Dissolved Oxygen, DO) 含量是决定PFC性能的一大关键因素,氧气浓度可以影响PFC装置中阴极处电子的消耗速率,以及相关含氧自由基的生成量。有无氧气存在也决定了阴极处发生还原反应的反应电位,从而影响PFC体系有机污染物降解效率以及产能发电性能。Ho等[110]在研究DO含量对于无膜PFC性能影响的研究中,对照了无曝气、低强度曝气、高强度曝气以及N2曝气等反应条件下PFC的性能,研究发现适度曝气能够增强PFC的性能,当曝气量从100 mL·min-1增加到200 mL·min-1时,DO含量增加了3.6%,污染降解速率常数是前者的2.02倍。原因是DO含量的提高,与电子结合生成H2O2以及· 等活性物质的含量升高,速率加快。但是,当曝气量从200 mL·min-1提高到1000 mL·min-1时,DO含量反而从8.15 mg L-1下降为8.01 mg·L-1,PFC的性能随之下降。
污染物的降解效率受到多种因素的影响,选择合适的DO含量可以有效提高PFC的性能,并且降低运行成本。Yao等[111]研究了PFC中DO含量对底物氧氟沙星降解效果和体系产能效率的影响。实验对照了不同DO含量下,氧氟沙星的降解率和反应速率,发现DO对氧氟沙星的降解具有促进作用,但是并不明显。通过分析原因主要有两方面:第一,在缺氧条件下,体系中的电子数量占优势,电子与底物之间的反应比·OH与底物之间的反应速率高10倍;第二,氧气的存在也会干扰原有的底物的自敏化光降解过程,造成活性物质之间的淬灭。实验还探究了不同DO含量对PFC的开路电压和最大功率密度的影响,DO为9 mg·L-1时,其Voc和JVmax分别是DO为0 mg·L-1时的1.7倍和5.4倍。另外,DO存在情况下,电路的Jsc是无氧条件下的1.6倍,这表明了DO提高了两电极之间的电势,O2作为电子受体,有效地利用了光生电子,提高了载流子的分离率。根据实际运行体系,对于不同的反应以及处理不同底物的PFC装置,选择合适的氧含量非常重要。
4.4.2 电解液/pH值
PFC系统中电解液的主要作用是传导离子,降低体系的传质阻力。常见的电解液有NaOH[82]等碱溶液、H2SO4[83]等酸溶液,或者Na2SO4[31]、NaCl[63]以及磷酸缓冲液[112]等盐溶液。电解质的类型、浓度和pH值等均会影响PFC性能。Li等[112]探究了不同电解液浓度 (0~0.5mol·L-1) 对TiO2-Ti阳极、Cu2O-Cu阴极构成的PFC体系性能的影响。该体系中磷酸盐缓冲液作为电解液,具有增强导电性和维持pH值稳定的作用。电解液浓度从0mol·L-1升高至0.2mol·L-1,Jsc和JVmax分别由约0.025mA·cm-2、0.15mW·cm-2提升到0.175mA·cm-2、0.225mW·cm-2。当电解液浓度进一步增加至0.5mol·L-1,二者数值保持不变。这是由于电解液浓度较小时,体系溶液的电导率影响着PFC的能效;而当电解液浓度超过临界值时,体系电子空穴对的激发以及分离率成为了影响PFC性能的决定性因素。此外,电解液中的许多离子还在光电催化过程中会参与到体系的氧化还原反应之中,例如Cl-能在PFC体系中能生成活性氯自由基[63],Zhou等[113]发现,当体系中含有50mmol·L-1的Cl-时,2 h内体系中总氮去除率从11.02%提升到94.93%。溶液的pH值对PFC性能影响尤为明显,诸多半导体材料在不同pH值下表现出截然不同的催化活性,不同电极材料降解污染物的最适pH范围也有所区别,如WO3在碱性环境中不稳定,Fe2O3易溶于酸性溶液。此外,溶液pH值也决定了PFC体系中阴极还原反应的反应电位,酸性环境有利于提高阴极的ORR反应的电位。而碱性环境下有利于阳极空穴的消耗。Chen等[114]通过探究KOH电解液pH变化对PFC性能的影响规律,发现电解液浓度越高,装置性能越佳。主要有以下三方面原因:(1) 高OH-浓度提高了空穴的利用率,促进了·OH生成,有效提高污染物的降解效果;(2) 高OH-浓度增加了阴极的氧化还原电位,从而提高了两电极之间的电位差;(3) 高OH-浓度提高了体系导电率和离子的传导效果。因此,根据所研究的体系选择合适的电解质以及pH有助于构建高效稳定的PFC体系。
4.4.3 底物
底物也称为PFC装置的“燃料”,底物的种类和浓度不同,PFC的性能也会有所差异。Chen等[114]考察了底物甲醇浓度对PFC性能的影响,结果表明:甲醇浓度提高了体系的传质效率,增加了空穴利用率,促进电子-空穴有效分离。当甲醇浓度从0.05mol·L-1升高至4.0mol·L-1,Jsc和JVmax分别由约0.52mA·cm-2、0.37mW·cm-2提升到0.65mA·cm-2、0.48mW·cm-2。Ho等[115]研究了不同底物分子结构与PFC性能之间的联系。在ZnO-Zn光阳极、Pt-C阴极构成的单光电极PFC系统中,考察了活性绿19 (RG19)、酸性橙7 (AO7)、亚甲基蓝 (MB) 三种有机染料作为目标底物时PFC体系中污染物降解效果和发电性能。由于不同污染物的分子结构及其理化性质存在较大差异,三种污染物的处理效果也各有差异。RG19具有供电子基团,分子结构中存在磺酸基,为阴离子染料,导致其更容易被氧化,ZnO表面带正电荷,静电吸引作用提高了RG19分子在电极材料表面的吸附性能;AO7因其在紫外光照射下的光敏化效果,在紫外光条件下去除性能更高;而可见光条件下,MB的脱色反应速率常数是紫外光条件下的5倍,这是由于可见光条件下,MB分子能够产生自敏化现象,从而促进体系产生更多自由基。
4.4.4 离子交换膜
离子交换膜在PFC装置中起到隔开阴极和阳极并允许特定离子通过的作用,通常包括质子交换膜和阴离子交换膜[116]。离子交换膜可以避免阳极发生的氧化反应与阴极发生的还原反应相互干扰,提高体系中自由基与载流子的利用率;还可以通过在阳极设置碱性环境,阴极营造酸性氛围来提高两极之间电势差,从而提升电子的传递效率和Voc。
Chen等[117]设计了CdS-ZnS量子点敏化TiO2 (CdS-ZnS/TiO2) 为阳极、铂碳涂覆碳纸为阴极的微流体PFC装置,两电极被离子交换膜隔开。该电池工作过程中,模型污染物通过蛇形通道运输到光电阳极表面,透过通道的光子能够激发阳极产生电子和空穴,电解质为KOH,h+可以与OH-反应生成·OH,继而氧化降解有机物;电子通过外部回路传递至阴极,与O2、H2O作用生成OH-,OH-又通过离子交换膜回到阳极形成循环,以达到持续降解污染物和产能发电的效果。在该PFC装置中,阴离子交换膜增强了OH-的运输效果,理论上阳极消耗的OH-可以通过阴极的还原反应得到补偿;另外该交换膜也减少了底物乙醇在两极之间交叉扩散。Jia等[87]利用全氟离子交换膜将转盘式PFC两电极隔开,阳极反应池为2mol·L-1 NaOH碱性溶液环境,阴极反应池为1mol·L-1 H2SO4强酸性溶液环境。全氟离子交换膜只允许氢离子单向通过,保证了两电极酸碱性互不干扰。阳极的碱性环境,有利于空穴转化为·OH;酸性环境可以提高阴极的电势,有利于析氢反应发生。两电极之间的电势差增大,PFC的性能得到了有效提高。
离子交换膜是双室电化学反应池装置最重要的组件之一,对于提高阴极还原反应效率起着重要作用,但存在着价格昂贵、被污染后性能下降以及需要定期更换等问题。因此,研发低成本、高稳定性的离子交换膜对于PFC装置实际应用以及推广具有重要的意义。
4.4.5 活性氧及活性自由基
通过在体系中外加目标离子,或者调控体系溶解氧、pH等操作条件,可以有效提升特定活性物种的生成量,从而有效提升体系中污染物的除去效果,提高PFC体系的各项综合性能。在体系中投加Fe2+等手段,可以将体系中如H2O2物种转化为氧化性较强的·OH。Zhou等[120]构建了阳极为TiO2/Ti,阴极为AQS/PPy-CF的PFC体系。阳极处半导体在光照下产生的h+可以与水分子反应产生·OH,阴极电极表面吸附的O2分子可以原位还原为· ,· 可以发挥降解有机污染物作用,也可以转化为H2O2以及·OH等活性物种。此外,· 也较好促进了Fe(Ⅱ)和Fe(Ⅲ)之间的转化,被认为是该体系中最重要的活性物种。除此之外,他们还构建了Fenton/PFC体系[71],有效地在体系中引入·OH、·O2、· 以及H2O2等活性基团,提高了体系中活性物种种类与数量。
5 PFC的应用
5.1 有机污染物的降解
PFC在实际废水处理中也表现优异。Zhou等[15]利用STNA作为阳极的PFC体系处理多种含有高浓度COD的实际废水,例如制药废水、石油开采废水、染料废水、化工厂废水等,不同废水条件下PFC体系均得到了较大的Voc以及Jsc,PFC在处理印染废水时得到的Voc可达1.53 V,Jsc可达1.21mA·cm-2,JVmax可达0.50mW·cm-2。TiO2/ZnO/Zn阳极、TiO2/CuO/Cu阴极构成的双光电极PFC系统[122],可以有效处理高COD和BOD的棕榈油厂实际废水。浓度为100 mg L-1的废水在最佳反应条件下,COD去除率高达89%,PFC系统的Voc为1.17 V,Jsc为0.2652mA·cm-2,JVmax为0.0734mW·cm-2,体系表现出了长时间运行稳定性与耐用性。利用Ag/TiO2阳极构建的流动相PFC体系在模拟太阳光照射下,处理实际啤酒厂废水,COD去除率为14.8% (532 mg·L-1),输出功率可达1.85 W·m-2[123]。
5.2 析氢
氢气具有高能量密度,使用过程中无二次污染的优点,是目前最引人注目的清洁能源之一。在合适的条件下,PFC体系中自发移动到阴极的电子通过还原质子生成氢气。Li等[31]发现,在WO3阳极、Pt阴极的PFC体系中,可以实现阳极氧化处理有机污染物、阴极还原析氢的双重效益。光生空穴的高效捕获和利用能够明显提升阴极的析氢效果。Yang等[124]为了实现阴极连续析氢,在PFC系统的TiO2阳极上并联了超级电容。该电容可以储存阳极材料光照下产生的电子,在黑暗条件下释放至阴极发生产氢反应。PFC系统在初始光照24 h内实现了底物乙二醇48%的去除率,光照和暗态条件下该系统的析氢产量分别可达32 μmol·L-1以及13 μmol·L-1。与电催化析氢过程类似,开发高效、低廉的电极材料也是解决PFC中析氢过程实际应用的关键问题之一。
5.3 重金属还原
环境水体中的重金属离子,如Cu2+[125]、Hg2+、Cr6+[126]、Pb2+[127]、Cd2+等均可利用光生电子转化为金属单质或者低价无毒产物。不同的金属离子的还原反应电势有所差别,如Cu2+/Cu为0.34 V (vs NHE pH=0),Ag+/Ag为0.8 V (vs NHE pH=0),$\mathrm{Cr}_{2} \mathrm{O}_{7}{ }^{2-}$/Cr3+为1.33 V (vs NHE pH=0),所以不同的重金属离子在阴极的还原能力也会有所差异[128]。根据溶液中重金属离子种类以及含量,选择合适的电极以及溶液pH非常重要。此外,阴极表面的金属离子还原后会沉积在电极表面。如Zeng等[65]利用MoO3/ZnO/Zn阳极、Pt/CC阴极构成的PFC装置处理浓度为40 ppm苯酚溶液,太阳光下反应4 h,阳极底物的降解率为89.5%,与此同时,阴极处 (200 ppm Cu2+) 的处理效率高达93.6%。反应结束后对Pt/CC阴极电极表面进行元素分析,研究发现电极表面沉积了大量的Cu物种,后续研究表明这些Cu物种可以作为电极表面新的反应活性位点,促进了Cu2+离子还原反应以及ORR反应的发生。
6 结论与展望
PFC是集污染物降解与产电功能于一体的新型技术,具有非常广阔的应用前景。本综述从PFC的基本结构以及原理入手,对比了单光电极、双光电极PFC装置结构和工作原理的差异;从阳极、阴极材料优化、反应器结构设计以及反应条件调控四个方面较为详细地介绍了PFC性能调控以及优化的策略以及手段;总结了PFC装置在污染物降解、产氢以及重金属还原等领域的应用及研究现状,为该领域的后续研究以及工业化应用提供了一定的理论指导。
PFC可以实现污染物降解、产电、产氢以及重金属还原等多重效益。但是,PFC的工业化还需要从以下几个方面展开研究:(1) 选择制备工艺简单、价格低廉、运行稳定性高以及性能优越的电极材料;(2) 合理设计反应器结构,尽量减少溶液对光的反射,有效提高半导体电极表面光子的利用率;(3) 根据体系的电极材料以及应用场景,合理调控装置的操作环境以及条件。