





Contents
1 Introduction
2 Cs2CO3-assisted palladium-catalyzed functionalization of C—H
2.1 Cs2CO3-assisted palladium-catalyzed functionalization of C(sp2)—H
2.2 Cs2CO3-assisted palladium-catalyzed functionalization of C(sp3)—H
2.3 Cs2CO3-assisted palladium-catalyzed functionalization of C(sp)—H
3 Cs2CO3-assisted palladium-catalyzed functionalization of O—H
4 Cs2CO3-assisted palladium-catalyzed functionalization of N—H
5 Cs2CO3-assisted palladium-catalyzed functionalization of B—H
6 Conclusion and outlook
1 引言
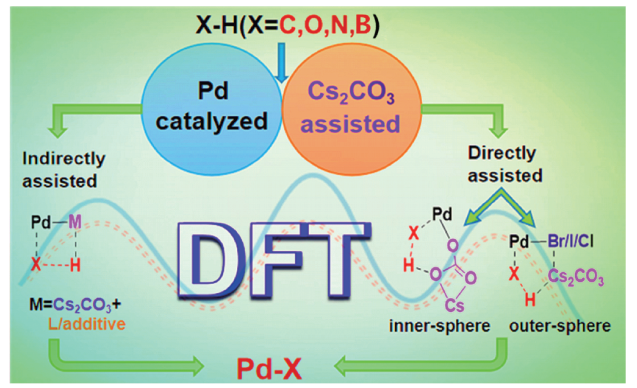
随着研究的深入,人们发现在钯催化X—H (X=C、O、N、B)官能团化反应中加入碱性添加剂如Cs2CO3可加快反应速率或提高产率[22⇓⇓⇓~26],并推测Cs2CO3加速反应的机制是由于其在醇、N,N-二甲基甲酰胺(DMF)、乙醚等有机溶剂中具有较好的溶解性[27,28]。但由于过渡金属d轨道的存在及其在催化反应中广泛参与作用的事实[29,30],使得Cs2CO3辅助钯催化X—H (X=C、O、N、B)官能团化反应的微观机理异常复杂。为了弄清楚该反应的动力学机制,部分实验研究者利用动力学同位素效应(Kinetic Isotope Effect, KIE)实验来预测反应的决速步,发现大多为X—H (X=C、O、N、B)键的活化步骤[31⇓~33]。然而,传统实验很难观察或探测到反应底物、钯催化剂与Cs2CO3之间的微观作用机制。为有效解决该问题,近年来,很多理论工作者采用量子化学方法对Cs2CO3辅助钯催化X—H (X=C、O、N、B)官能团化反应进行计算,其中密度泛函理论(Density Functional Theory, DFT)是使用最为广泛的方法[34⇓⇓⇓⇓~39]。他们通过对反应体系的计算模拟,获得反应路径上各驻点的结构和能量信息,并基于此揭示反应的微观机理和选择性调控机制等,为深入认识Cs2CO3辅助钯催化X—H (X=C、O、N、B)键活化的反应本质提供了重要帮助。目前,关于钯催化X—H (X=C、O、N、B)官能团化的理论研究成果有不少综述提及[40,41],并被国内外优秀期刊论文广泛引用[42⇓⇓⇓⇓⇓⇓⇓⇓~51],但尚未见有关Cs2CO3辅助钯催化X—H (X=C、O、N、B)官能团化反应的理论研究成果的全面总结与评述。
事实上,钯催化X—H (X=C、O、N、B)官能团化在过去十年内得到了长足发展[52],尤其是在Cs2CO3辅助钯催化X—H官能团化这一领域,不断有优秀的实验研究成果被报道[53⇓⇓⇓~57],也有不少理论研究者使用密度泛函理论对此类反应的微观机理、选择性产生根源及Cs2CO3作用机制等进行了计算研究[58⇓~60]。研究显示,Cs2CO3的络合能力很强,可与Cs2CO3自身、配体或添加剂等形成多聚体或络合物,以增强反应活性,降低反应能垒[61,62]。2019年,刘靖尧等[63]对Cs2CO3的单体、二聚体和四聚体进行了理论计算,发现四聚体最为稳定,以1/4(Cs2CO3)4作为能量参考零点更为合理。不过,在探讨Cs2CO3辅助钯催化X—H (X=C、O、N、B)官能团化的反应机理和反应选择性时,由于反应难易和反应选择性结果受能量零点选取的影响较小,且通常只有一个Cs2CO3分子能直接介入活化过程,多数作者仍以单分子Cs2CO3进行计算[58,64,65]。
2 Cs2CO3辅助钯催化C—H官能团化
C—H键是有机化合物中最为普遍的化学键,根据其所连接碳原子的杂化类型不同,可将C—H键区分为C(sp3)—H、C(sp2)—H、C(sp)—H等。C(sp3)—H主要是烷基C—H,C(sp2)—H包括芳基、烯基及醛基C—H等,C(sp)—H主要为炔基C—H。目前,关于Cs2CO3辅助钯催化C(sp2)—H官能团化的研究最为透彻,而Cs2CO3辅助钯催化烷基C(sp3)—H官能团化和炔基C(sp)—H官能团化的理论研究报道相对较少。本节将依次对Cs2CO3辅助钯催化C(sp2)—H、C(sp3)—H和C(sp)—H官能团化的理论研究成果进行总结与讨论。
2.1 Cs2CO3辅助钯催化C(sp2)—H官能团化
根据Cs2CO3辅助钯催化C(sp2)—H键活化的方式不同,可将其分为Cs2CO3直接辅助和Cs2CO3间接辅助两类。其中,Cs2CO3直接辅助钯催化C(sp2)—H活化是指:当只有Cs2CO3作为反应添加剂时,Cs2CO3直接辅助底物的C(sp2)—H活化。Cs2CO3间接辅助是指:当其他添加剂或配体与Cs2CO3共同辅助钯催化C(sp2)—H活化反应时,Cs2CO3通过协助其他添加剂或配体中的O—H活化来最终实现对C(sp2)—H的活化。
2.1.1 Cs2CO3直接辅助钯催化C(sp2)—H活化
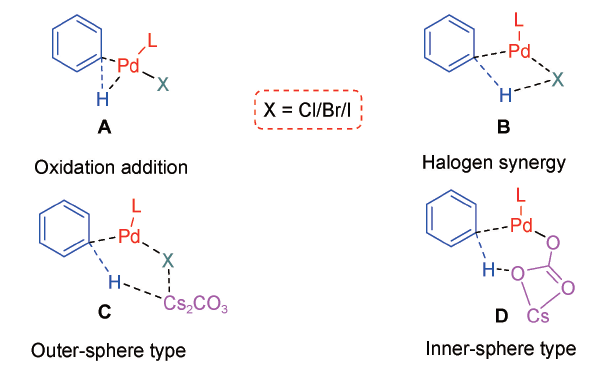
近十年,关于Cs2CO3直接辅助钯催化芳基C(sp2)—H活化的理论研究时有报道[58,63,70⇓~72],主要涉及通过偶联[73]、螺环化[74]或稠环化过程[75]获得复杂功能分子的反应。常见的芳基C(sp2)—H活化机制可分为氧化加成型(A)、卤素协同型(B)、外碱辅助型(C)和内碱辅助型(D)等(图式1 )[9,76]。其中,外碱辅助型一般对应碱与卤素配位,辅助C—H活化后脱去CsX·CsHCO3团簇、或脱去CsX与CsHCO3;内碱辅助型通常为碳酸盐取代卤素/配体离子、与钯配位的同时脱去CsX(X为卤素/配体离子),随后CsC 与钯配位辅助C—H活化并脱去CsHCO3。本小节主要涉及Cs2CO3直接辅助的外碱辅助型(C)和内碱辅助型(D)两类。
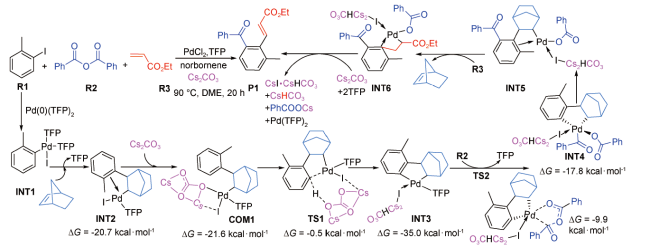
2017年,毕思玮等在PCM(DME)/M06/ 6-311++G(d,p)-SDD(Pd,P,I,Cs)//B3LYP/6-31G(d)-LANL2DZ(Pd,P,I,Cs)水平上,对许鹏飞等报道的钯催化邻甲基碘苯、苯甲酸酐和丙烯酸乙酯发生交叉偶联生成芳基酮的反应[77]进行了DFT计算[70]。结果显示:该反应主要经历氧化加成、降冰片烯插入、C—H活化、碳碳成键、降冰片烯脱去和还原消除等过程(图式2 )。在芳基C(sp2)—H的活化过程中(INT2→INT3),由于Cs2CO3以外碱辅助形式进行协助,反应跨越过渡态TS1的Gibbs自由能垒仅为21.1 kcal·mol-1,比决速步过渡态TS2的能垒(25.1 kcal·mol-1)低4.0 kcal·mol-1,说明Cs2CO3的辅助对该反应的顺利进行具有重要意义。
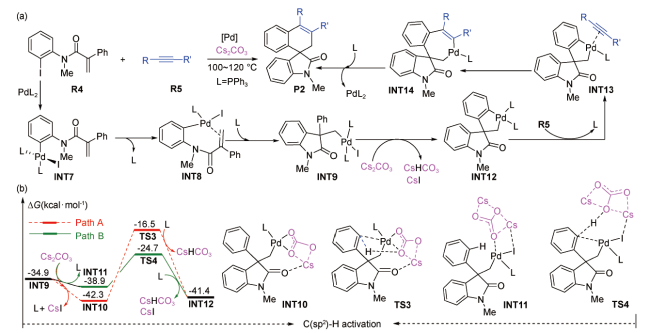
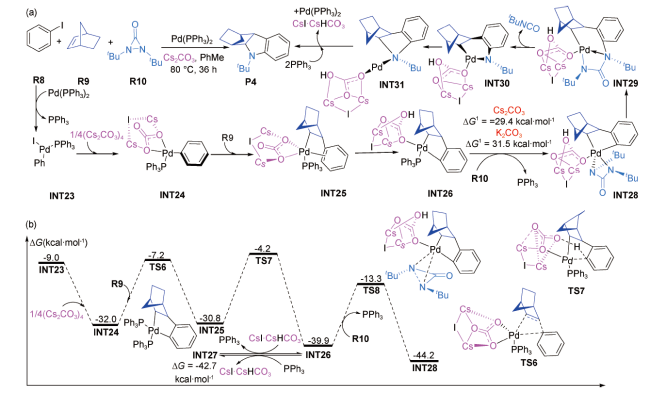
为深入探讨钯催化C—H键活化反应过程中的外碱和内碱辅助机制,2018年,Lautens等在IEFPCM(Tol)/M06L/def2-TZVPP//B3LYP/6-31G(d,p)-LANL2DZ(Pd,Cs,I)水平上,对钯催化丙烯酰胺螺环化构建含氮螺杂环的反应机理进行了DFT研究[58]。计算结果显示:催化剂PdL2先与底物R4发生氧化加成转化为中间体INT7(图式3 a),之后中间体INT8经C—C成键形成五元环中间体INT9。INT9中的C(sp2)—H在Cs2CO3辅助下发生活化,经Path A和Path B两条可能路径转化为螺环化中间体INT12(图式3 b):其中Path A对应Cs2CO3与碘配体交换脱去CsI后、以内辅助形式协助芳基C(sp2)—H活化的过程,需克服25.8 kcal·mol-1(TS3)的Gibbs自由能垒;Path B对应Cs2CO3以外辅助形式协助芳基C(sp2)—H活化脱去CsHCO3和CsI的过程,仅需跨越14.2 kcal·mol-1(TS4)的Gibbs自由能垒,说明该C—H活化反应更倾向于以外碱辅助机制进行。最后,底物R5与中间体INT12作用,经配体交换、炔烃插入和还原消除过程生成P2产物(图式3 a)。该理论研究结果对后续深入探讨Cs2CO3辅助钯催化C(sp2)—H活化的外碱和内碱辅助机制具有重要的奠基意义。
2019年,Koley等在SMD(Tol)/B3LYP-D3(BJ) /6-311++G**-LANL2TZ(f)(Pd,Cs)//B3LYP-D3/6-31+G**-LANL2DZ(Pd,Cs)水平上,对钯催化4-甲氧基溴苯和多氟代烃羰基的偶联机理进行了DFT研究[71]。计算结果显示:反应经历了CO配体交换、氧化加成、CO迁移插入、C(sp2)—H活化、C—C成键和还原消除过程(图式4 a)。Cs2CO3以内辅助形式协助芳基C(sp2)—H活化的过程(INT19→TS5→INT20)为整个反应的决速步(图式4 b),需要跨越的Gibbs自由能垒为20.2 kcal·mol-1。他们还对K2CO3和Na2CO3辅助的反应进行了对比研究,发现其反应机理与Cs2CO3辅助的反应相似,但决速步的Gibbs自由能垒均比Cs2CO3辅助的高(图式4 a),说明Cs2CO3对该反应的辅助效果更好。同年,刘靖尧等在PCM(Tol)/M06L/6-311+G**-LANL2DZ(Pd,P,I,Cs)//M06L/6-31G(d,p)-LANL2DZ(Pd,P,I,Cs)水平上,对钯催化碘苯、降冰片烯与二叔丁基二氮杂环丙啶酮的反应机理进行了DFT研究[63]。计算结果显示:反应经历了C—I氧化加成、CsI·CsHCO3团簇形成、降冰片烯插入、芳基C(sp2)—H活化、N—N氧化加成、C(sp2)-N形成、C(sp3)-N形成和还原消除过程(图式5 a)。反应中Cs2CO3以外辅助形式协助芳基C(sp2)—H活化(INT25→TS7→INT26)(图式5 b),相应的Gibbs自由能垒为26.6 kcal·mol-1,比决速步过渡态TS8的能垒①(①该文献中,作者计算决速步能垒选用的是中间体INT27与过渡态TS8之间的差值。)(29.4 kcal·mol-1)低2.8 kcal·mol-1,可见,Cs2CO3的辅助使C(sp2)—H活化变得更加容易。他们还对K2CO3和Cs2CO3辅助的反应进行了对比研究,发现K2CO3辅助的决速步Gibbs自由能垒比Cs2CO3辅助的决速步Gibbs自由能垒(29.4 kcal·mol-1)高2.1 kcal·mol-1,说明Cs2CO3对该Heck反应的辅助效果更好。
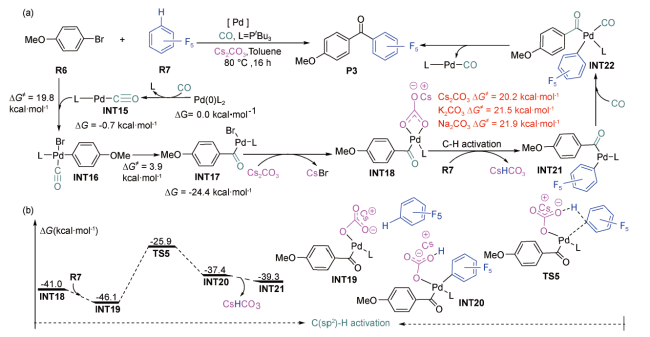
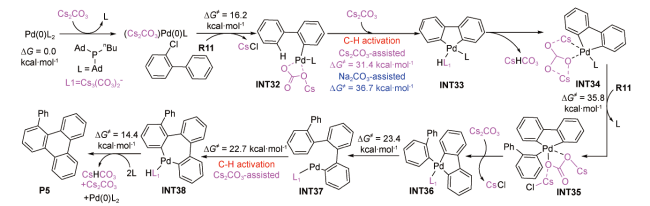
受上述Koley等的研究结果[71]启发,2020年,Itami等在IEFPCM(DCM)/B3LYP-D3(BJ)/6-311 +G(d,p)-SDD(Pd,Cs)//B3LYP-D3(BJ)/6-31G(d,p)-LANL2DZ(Pd,Cs)水平上,对钯催化2-氯联苯制备稠环化合物的反应机理进行了DFT研究[72]。计算结果显示:反应经历了Cs2CO3与膦配体交换、C—Cl裂解、芳基C(sp2)—H活化、C—Cl裂解、C—C成键、芳基C(sp2)—H活化、C—C成键和还原消除过程(图式6 )。Cs2CO3在整个反应过程中多次参与作用,并以内辅助形式两次协助芳基C(sp2)—H活化,相应的Gibbs自由能垒依次为31.4和22.7 kcal·mol-1,均低于C—Cl裂解决速步的能垒(INT34→INT35,35.8 kcal·mol-1)。此外,Cs2CO3第一次辅助芳基C(sp2)—H活化的Gibbs自由能垒较Na2CO3辅助的能垒(36.7 kcal·mol-1)低5.3 kcal·mol-1,说明Cs2CO3更有利于提升C(sp2)—H活化的反应速率。
同年,Poater等在PCM(Tol)/M06/cc-pVTZ- SDD(Pd)//BP86-D3/SVP-SDD(Pd)水平上,对钯催化苯基三异丙基甲硅烷基乙炔基醚与新戊酸肉桂酯的反应机理进行了DFT研究[78]。计算结果显示:该反应主要经历了烯基配位、Cs2CO3配位脱PivOCs、芳基C(sp2)—H活化、C—C成键、H迁移、C—C成键、烷基C(sp3)—H活化和还原消除等过程(图式7 a)。在中间体INT39转化为中间体INT44的过程中(图式7 b),Cs2CO3同样以内辅助形式协助芳基C(sp2)—H活化,是整个反应的决速步,相应的Gibbs自由能垒为35.9 kcal·mol-1(TS10),在140℃的实验条件下略显偏高。
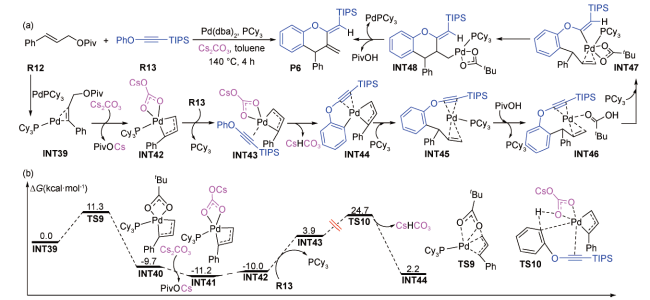
图式7 Cs2CO3辅助钯催化苯基三异丙基甲硅烷基乙炔基醚与新戊酸肉桂酯双C—H裂解的环化反应机理(a)及其芳基C(sp2)—H活化的势能剖面图(b)[78]Scheme 7 (a) Reaction mechanism for Cs2CO3-assisted palladium-catalyzed double C—H cracking and cyclization of phenyl triisopropylsilyl ethynyl ether with cinnamyl pivalate, and (b) potential energy profiles for aryl C(sp2)—H activation[78] |
综合上述理论研究结果,在钯催化芳基C(sp2)—H活化过程中,Cs2CO3以内辅助形式参与作用的情况居多,但也有少部分反应中Cs2CO3以外辅助形式参与作用的路径在能量上更有利。因此,对不同的反应体系开展理论研究时,应该全面考虑Cs2CO3以外辅助和内辅助形式参与作用的可能性。
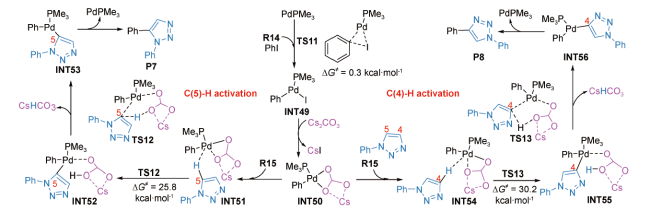
如2015年,Patil等以N-芳基-1,2,3-三唑和碘苯为底物、Pd(OAc)2为催化剂、Cs2CO3为碱,在100℃条件下反应制备得到了N-芳基-1,2,3-三唑衍生物[86],并在M06L/6-311G**-LANL2DZ(Pd,Cs)水平上对该反应的机理进行了DFT研究。结果显示:Pd(0)与碘苯发生氧化加成转化为中间体INT49仅需克服0.3 kcal·mol-1的Gibbs自由能垒(图式8 ),接着Cs2CO3与中间体INT49作用,通过配体交换转化为中间体INT50。底物R15进入后,经协同金属-去质子化(Cooperative Metallization-Deprotonation,CMD)实现烯基C(sp2)—H活化。其中,Cs2CO3以内辅助形式协助C(5)—H或C(4)—H活化的Gibbs自由能垒分别为25.8(TS12)和30.2 kcal·mol-1(TS13),说明反应更倾向于形成C(5)—H活化的产物P7,反应的区域选择性由此产生。该工作不但为制备N-芳基-1,2,3-三唑衍生物开拓了一种新方法,也为人们深入认识Cs2CO3辅助钯催化烯基C(sp2)—H活化的反应机制提供了新视角;理论计算所揭示的区域选择性产生根源对实验上设计方案、以获得具有特定区域选择性的产物具有积极的指导意义。
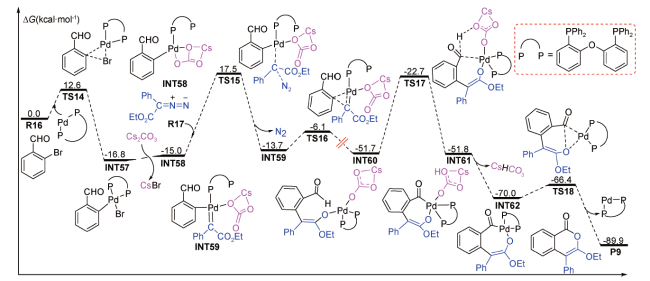
在Cs2CO3辅助钯催化芳基C(sp2)—H和烯基C(sp2)—H官能团化取得显著成效的背景下,Cs2CO3辅助钯催化醛基C(sp2)—H官能团化也引起了人们的注意。2018年,黄学良等报道了钯催化芳基重氮酯与邻溴苯甲醛的分子间酰化反应[65],这是关于Cs2CO3辅助钯催化醛基C(sp2)—H活化的首个反应实例。他们在SMD(Tol)/M06/def2-TZVP//B3LYP/ 6-31G(d)-LANL2DZ(Pd,Br,Cs)水平上对该反应进行了DFT计算。结果显示:邻溴苯甲醛与钯催化剂发生氧化加成需克服12.6 kcal·mol-1的Gibbs自由能垒(TS14, 图式9 ),之后底物R17与中间体INT58作用、经过渡态TS15形成卡宾中间体INT59,该过程是整个反应的决速步,需跨越34.3 kcal·mol-1的Gibbs自由能垒。之后,Cs2CO3以内辅助形式协助中间体INT60中的醛基C(sp2)—H活化,相应的Gibbs自由能垒为29.0 kcal·mol-1(TS17),比决速步能垒低5.3 kcal·mol-1,说明Cs2CO3辅助使醛基C(sp2)—H活化变得更加容易。该工作对人们进一步探究醛基的C(sp2)—H活化反应机制有奠基意义。
2.1.2 Cs2CO3间接辅助钯催化C(sp2)—H活化
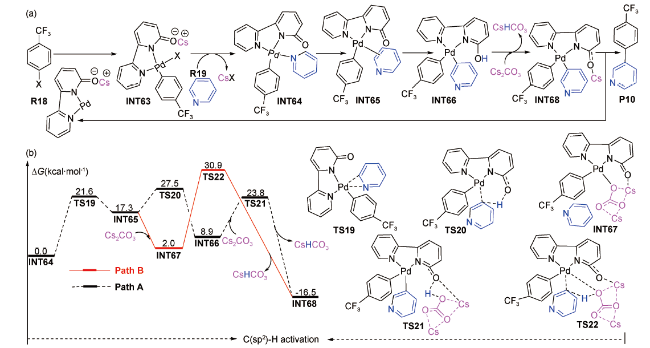
与上述Cs2CO3直接辅助钯催化C(sp2)—H活化方式不同,Cs2CO3辅助钯催化C(sp2)—H活化也可采用间接方式。如2018年,AlbÉniz等使用DFT理论,在SMD(pyridine)/M06/6-311++G(d,p)-LANL2TZ(f)(Pd,Cs,Br,I)//M06/6-31+G(d)-LANL2TZ(f)(Pd,Cs,Br,I)水平上,对钯催化吡啶与对-三氟甲基卤代芳基化合物的芳基化偶联反应中[87]所涉及到的Cs2CO3辅助钯催化C(sp2)—H活化过程进行了理论计算。结果显示:反应经历了氧化加成、配体交换、C(sp2)—H活化和还原消除等过程(图式10 a),其中C(sp2)—H活化存在Path A和Path B两条可能路径(图式10 b)。Path A对应配体2,2'-联吡啶-4-氧(bipy-4-O)先辅助C(sp2)—H活化的过程,需跨越27.5 kcal·mol-1的总Gibbs自由能垒(TS20)。形成中间体INT66后,Cs2CO3以外辅助形式协助配体的O—H活化,经过渡态TS21、克服14.9 kcal·mol-1的Gibbs自由能垒转化为中间体INT68。Path B对应Cs2CO3以外辅助形式直接协助吡啶环的C(sp2)—H活化的过程,经过渡态TS22、跨越30.9 kcal·mol-1的总Gibbs自由能垒转化为中间体INT68。上述结果表明该C(sp2)—H活化反应更倾向于以Path A的方式进行,对于人们深入认识Cs2CO3间接辅助钯催化C(sp2)—H活化的微观机制具有很好的启示作用。
2019年,徐利文等又报道了钯催化甲苯磺酰基保护的二芳基甲基胺羰基化制备4-苯基取代异喹啉酮的反应,产率可达87%[89]。他们在M06L/ 6-31G(d,p)-LANL2DZ(Pd,Cs)水平上对芳基C(sp2)—H活化机理进行了DFT研究,结果显示:Cs2CO3先辅助叔丁氧羰基-L-缬氨酸(Boc-L-Val-OH)发生O—H活化形成Boc-L-Val-OCs,并与Pd(Ⅱ)催化剂结合转化为中间体INT69 (图式11 )。
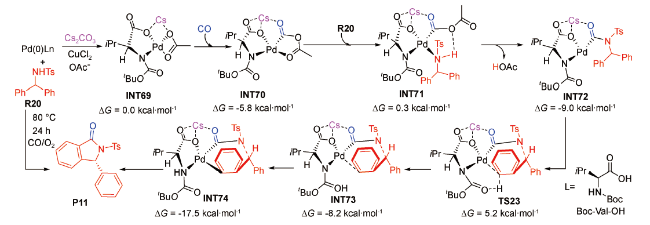
接着CO和底物R20相继进入反应体系,经C-N成键转化为中间体INT72。之后,Boc-L-Val-OCs以内辅助形式协助芳基C(sp2)—H活化,该过程为反应的决速步,相应的Gibbs自由能垒为14.2 kcal·mol-1(TS23)。最后,中间体INT74发生还原消除,形成产物P11。该理论研究结果对人们深入认识Cs2CO3间接辅助钯催化C(sp2)—H活化的微观机制同样具有重要的参考价值。
2.2 Cs2CO3辅助钯催化C(sp3)—H官能团化
2.2.1 Cs2CO3直接辅助钯催化C(sp3)—H活化
2018年,陈弓等报道了Cs2CO3辅助钯催化4-甲氧基碘苯与3-芳基丙酰胺经烷基C(sp3)—H活化制备偶联产物P12的反应[92](图式12 ),并在SMD (m-xylene)/ωB97XD/def2TZVPP//ωB97XD/6-31G(d)-LANL2DZ(Pd,Cs)水平上对该反应进行了DFT研究。计算结果显示:反应经历了氧化加成、配体交换、烷基C(sp3)—H活化和还原消除过程。其中Cs2CO3以内辅助形式协助烷基C(sp3)—H活化的过程为整个反应的决速步,仅需克服29.0 kcal·mol-1的Gibbs自由能垒(TS24),与反应在100℃时进行48 h得到95%的P12产物很好吻合。
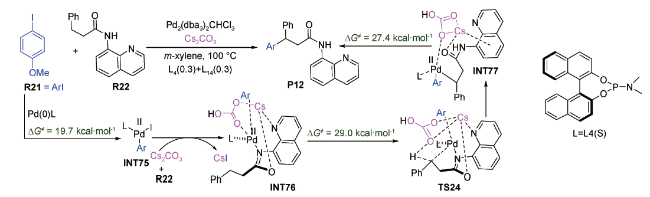
一年后,谢湖均等在CPCM(Tol)/M06/ 6-31G(d,p)-LANL2DZ(Pd,P,Br,Cs)水平上,对钯催化(Z)-2-溴乙烯基苯与endo-N-(对苯甲基)-降冰片烯琥珀酰亚胺反应制备环丙烷骨架的反应进行了DFT研究[93]。计算结果显示(图式13 ):反应经历了氧化加成、底物R24迁移插入、Cs2CO3和CsC 与溴配体交换、烷基C(sp3)—H活化、分子内烯烃插入、β—H消除和还原消除等过程。其中,Cs2CO3以内辅助形式协助烷基C(sp3)—H活化的Gibbs自由能垒为8.9 kcal·mol-1(TS25),比决速步过渡态TS26的能垒(28.5 kcal·mol-1)低19.6 kcal·mol-1;而无Cs2CO3辅助时,经中间体INT79转化为中间体INT87需要吸收86.7 kcal·mol-1的Gibbs自由能,说明Cs2CO3辅助是该钯催化烷基C(sp3)—H活化反应得以实现的重要条件。
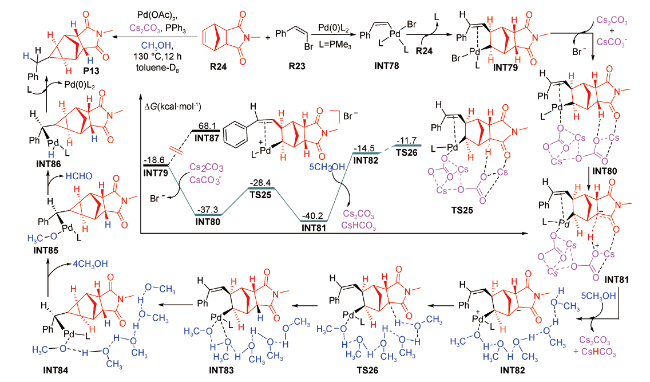
图式13 Cs2CO3辅助钯催化(Z)-2-溴乙烯基苯与endo-N-(对苯甲基)-降冰片烯琥珀酰亚胺[2+1]环加成的反应机理及其烷基C(sp3)—H活化的势能剖面图[93]Scheme 13 Reaction mechanism for Cs2CO3-assisted palladium-catalyzed [2+1] cycloaddition of (Z)-2-bromovinylbenzene with endo-N-(p-tolyl)-norbornene succinimide, and potential energy profiles for alkyl C(sp3)—H activation[93] |
基于前人对Cs2CO3辅助钯催化烷基C(sp3)—H活化的研究成果[92,93],2020年,陈弓等以3-苯基丙酰胺为底物、芳基碘化物为氧化剂,在100℃条件下制备得到了β-内酰胺产物(P14),产率可达92%[94]。他们通过在SMD(m-xylene)/M06/6-311+G(d,p)-SDD (Pd,I)//B3LYP-D3/6-31G(d)-SDD(Pd,I)水平上对该反应的化学选择性开展机理研究,发现反应经历了烷基C(sp3)—H活化、氧化加成、配体交换、C—C/C—N成键和还原消除过程(图式14 a)。Cs2CO3在反应中的作用为辅助烷基C(sp3)—H活化、后以HC 形式协助C—C/C—N发生还原消除。另外,他们分别以邻甲氧基碘苯(R26)和2-甲氧基-5-氯碘苯(R27)为氧化剂进行化学选择性研究(图式14 b),结果表明:以中间体INT89为零点(无HC 协助的路径)、发生C—C/C—N还原消除反应的能垒均高于以中间体INT90为零点(有HC 协助的路径)所对应的能垒,说明HC 协助反应可有效降低能垒。
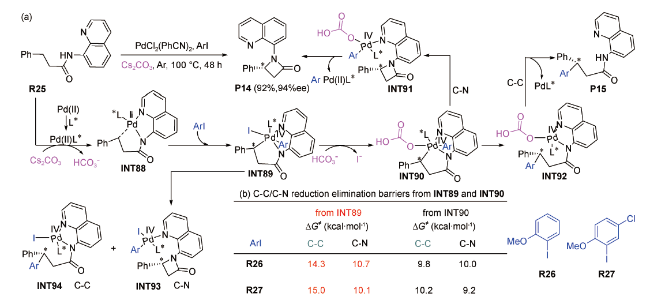
上述关于Cs2CO3直接辅助钯催化烷基C(sp3)—H活化的理论研究成果表明:Cs2CO3以内辅助形式参与反应可有效降低关键中间体的相对Gibbs自由能、以及烷基C(sp3)—H活化反应的能垒,从而使反应可在较为温和的实验条件下进行。这些理论研究成果对后续设计开发其他简便易行的烷基C(sp3)—H活化反应策略有一定的理论指导意义。
2.2.2 Cs2CO3间接辅助钯催化C(sp3)—H活化
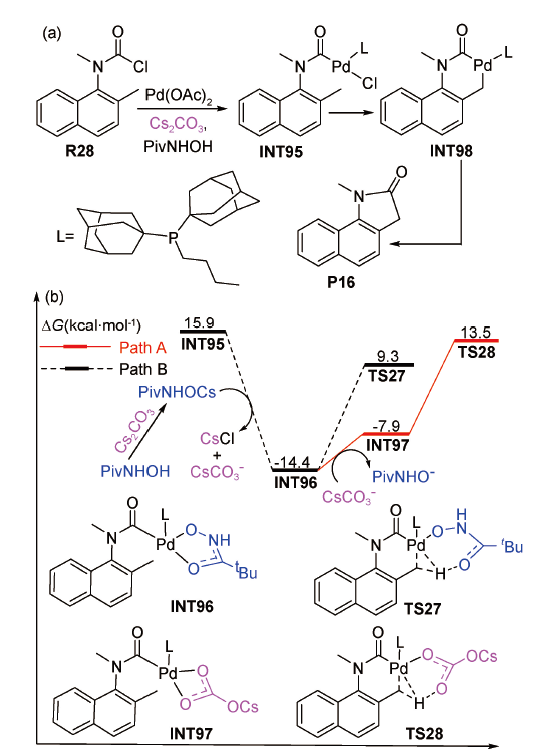
2013年,傅尧等在SMD(mesitylene)/M06/6-311 +G(d,p)-LANL2DZ(Pd,Cs)//B3LYP/6-31G(d)-LANL2DZ(Pd,Cs)水平上,对钯催化邻甲基萘氨基甲基甲酰氯的酰胺化反应机理进行了DFT计算[95]。结果显示:反应经历了氧化加成、烷基C(sp3)—H活化和还原消除等过程(图式15 a)。其中烷基C(sp3)—H活化有两条可能的反应路径:Path A和Path B(图式15 b)。Path A对应Cs2CO3以内辅助形式协助烷基C(sp3)—H活化的过程,需克服的Gibbs自由能垒为27.9 kcal·mol-1(TS28);Path B对应Cs2CO3先辅助新戊酰基羟胺(PivNHOH, Piv=pivaloyl)中的O—H活化生成PivNHOCs、之后再辅助烷基C(sp3)—H活化的过程,需跨越的Gibbs自由能垒为23.7 kcal·mol-1(TS27),比TS28低4.2 kcal·mol-1。上述结果表明:当PivNHOH和Cs2CO3共同辅助钯催化烷基C(sp3)—H活化反应时,Cs2CO3以间接方式辅助反应更有利。
一年后,Kündig等报道了钯催化N-异丙基氨基甲酸酯制备2,3-取代吲哚的反应[96],并在M06L/ 6-31G(d,p)-LANL2DZ(Pd,Br)水平上对该反应的机理进行了DFT计算。结果显示:反应经历了氧化加成、配体交换、烷基C(sp3)—H活化和还原消除过程(图式16 )。其中,烷基C(sp3)—H活化通过以下方式进行:Cs2CO3先以间接方式辅助新戊酸(tBuCOOH)发生O—H活化生成tBuCOOCs,之后,tBuCOOCs再以内辅助形式协助烷基C(sp3)—H活化,经过渡态TS29转化为中间体INT101或经过渡态TS30转化为中间体INT102。
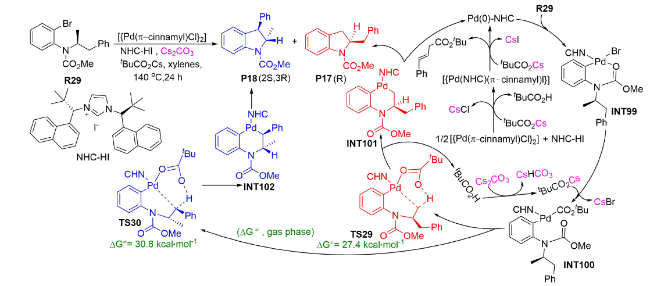
由于TS30的能垒比TS29高3.4 kcal·mol-1,反应更倾向于生成P17产物。该理论计算结果与实验所观察到的现象一致,较好地揭示了反应的化学选择性产生根源,为实验上开发新的钯催化C(sp3)—H选择性官能团化反应提供了有价值的理论参考。
基于Cs2CO3间接辅助钯催化C(sp3)—H活化机制的研究成果[95,96],梁永民等近期通过钯催化γ,δ-不饱和肟酯的Narasaka-Heck/C(sp3)—H活化制备得到了螺环化产物,产率可达75%[97]。他们进一步在PCM(Tol)/B3LYP/def2-SVP水平上对该反应进行了DFT计算。结果显示:反应经历了氧化加成、分子内Heck环化、C(sp3)—H活化和还原消除等过程(图式17 a)。其中,C(sp3)—H活化为整个反应的决速步,在配体C6F5C 的辅助下,需要跨越的Gibbs自由能垒为31.9 kcal·mol-1(TS31, 图式17 b),该能垒在150℃的实验温度下较为合理。随后的中间体INT105转化为五元螺钯环中间体INT106的过程为无位垒过程,该过程中Cs2CO3辅助C6F5CO2H中的O—H活化、促进C6F5C 从体系中脱去,并释放出27.3 kcal·mol-1的Gibbs自由能。
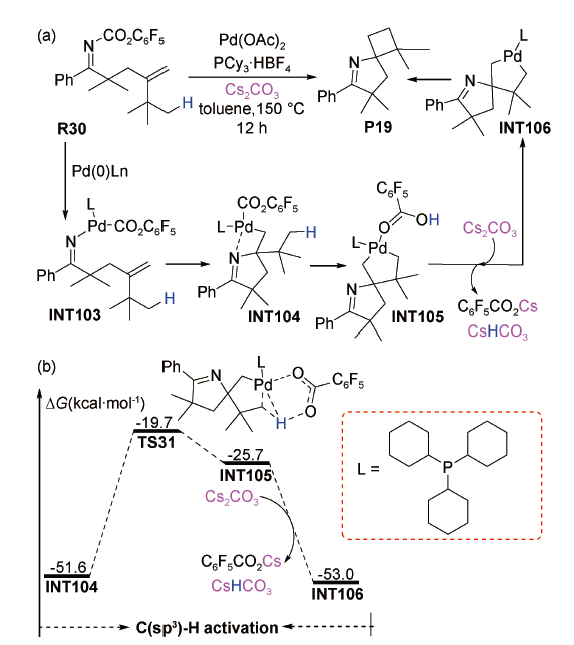
2.3 Cs2CO3辅助钯催化C(sp)—H官能团化
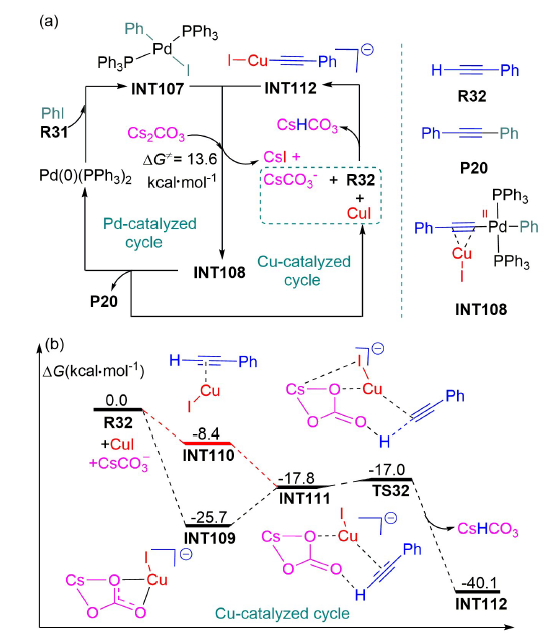
与Cs2CO3辅助钯催化C(sp2)—H和C(sp3)—H官能团化反应相比,关于Cs2CO3辅助钯催化炔基C(sp)—H官能团化反应的DFT理论研究报道明显偏少。2017年,罗一等在SMD(DMF)/M06/6-311+G(d,p)-SDD (Pd,Cu,I,Cs)//B3LYP/6-31+G*-LANL2DZ(Pd,Cu,I,Cs)水平上,对苯乙炔和碘苯发生Sonogashira交叉偶联反应的机理进行了DFT研究[98]。结果显示:铜催化和钯催化为两个相对独立又紧密相连的过程。在钯催化循环中,反应经历了氧化加成、炔烃插入和还原消除过程(图式18 a)。同时,钯催化循环中生成的CsC 通过与铜配位而参与到铜催化循环中(图式18 b),使CuI转化为中间体INT109,该中间体的能量比CuI与炔烃配位形成的中间体INT110低17.3 kcal·mol-1。之后,CsC 以内辅助形式协助炔基C(sp)—H活化,相应的Gibbs自由能垒低至8.7 kcal·mol-1(TS32),从而使中间体INT109经INT111和过渡态TS32快速转化为中间体INT112并参与到钯催化循环中。理论计算结果表明:该反应中Cs2CO3对炔基C(sp)—H活化以及两个催化循环的实现都起到了不可或缺的作用。
3 Cs2CO3辅助钯催化O—H官能团化
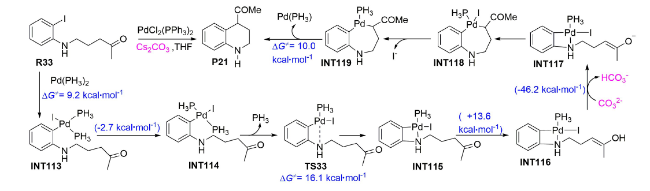
除Cs2CO3辅助钯催化C—H官能团化外,Cs2CO3辅助钯催化O—H官能团化反应也受到实验和理论化学研究者的极大关注[99⇓⇓~102]。如2011年,Fernández等在PCM(THF)/B3LYP/def2-SVP水平上对钯催化γ-(2-碘代苯胺基)酮的分子内偶联反应机理进行了DFT研究[103]。结果显示:底物R33与钯催化剂发生氧化加成(图式19 )、跨越9.2 kcal·mol-1的Gibbs自由能垒形成中间体INT113,后经中间体INT114实现Pd—N成键,转化为四元钯环中间体INT115,该步需跨越16.1 kcal·mol-1的Gibbs自由能垒(TS33)。之后,中间体INT116在Cs2CO3辅助下发生O—H活化转化为中间体INT117,并放出46.2 kcal·mol-1的Gibbs自由能,该步所放出热量对于驱动P21产物的形成起着至关重要的作用。
为进一步阐明Cs2CO3辅助钯催化O—H活化的反应机制,2016年,张松林等在SMD(Tol)/M06/ 6-311+G(d,p)-SDD(Pd)-LANL2DZ(Cs)//B3LYP/6-31G(d)-LANL2DZ(Pd,P,Cs)水平上对钯催化溴苯与4-甲基-4-羟基-2-戊酮发生交叉偶联制备α-芳基酮的反应机理进行了DFT研究[104]。计算结果显示:反应经历了氧化加成、O—H活化、C—C裂解和还原消除过程 (图式20 a)。其中,中间体INT120转化为INT121的O—H活化过程仅需吸收11.9 kcal·mol-1的Gibbs自由能(图式20 b),说明该O—H活化过程由于Cs2CO3的辅助而变得更加容易。
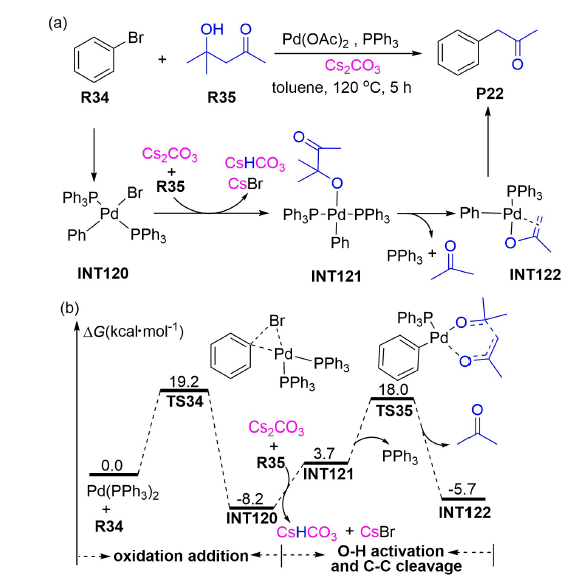
上述Fernández等和张松林等[103,104]的理论研究结果都表明Cs2CO3辅助能使钯催化O—H活化反应变得更加容易,但并未给出Cs2CO3参与作用的详细反应机制。2019年,周永贵等报道了钯催化1-溴-2-(1-苯基乙烯基)苯与1-苯基环丁醇反应制备交叉偶联产物P23(产率92%)的反应[88],并在B3LYP/6-311++ G(2df,2p)//SMD(Tol)/M05-2X/6-31G(d)-SDD//B3LYP/6-31+G(d)-SDD水平上对该反应的微观机理进行了DFT研究。计算结果显示:底物R36与钯催化剂发生氧化加成转化为中间体INT123之后(图式21 ),CsC 以外辅助形式、经过渡态TS36协助O—H活化,该过程仅需克服5.4 kcal·mol-1的Gibbs自由能垒。
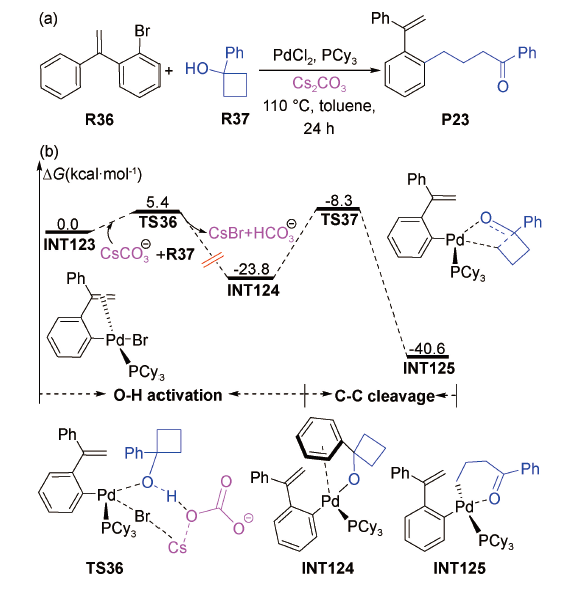
接着,中间体INT124发生C—C裂解开环,跨越15.5 kcal·mol-1(TS37)的Gibbs自由能垒转化为中间体INT125,并释放出大量的热。最后,中间体INT125发生还原消除形成产物P23。从图中可以看出,该反应中Cs2CO3的辅助对于降低钯催化O—H活化能垒和保证反应顺利进行都起到了关键作用。
在上述几个钯催化O—H活化反应中,由于有Cs2CO3的辅助,其O—H活化过程所涉及的的关键中间体或过渡态的相对能量得到有效降低,相应的反应能垒也明显下降,从而保证了反应的顺利发生。
4 Cs2CO3辅助钯催化N—H官能团化
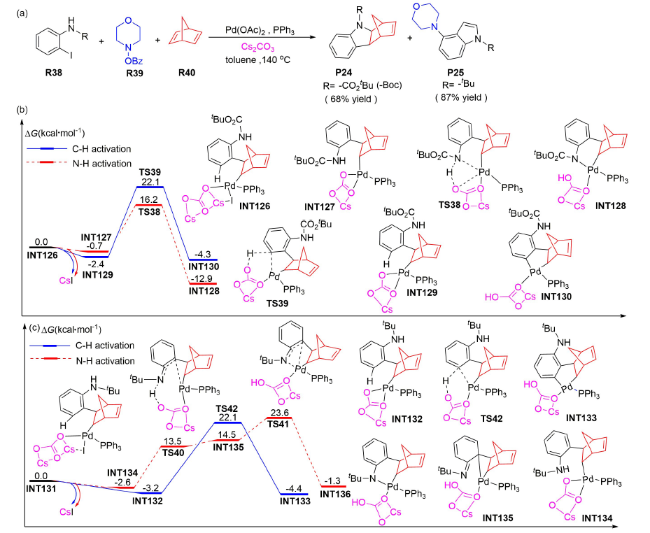
如2019年,梁永民等报道了钯催化邻碘苯胺、N-苯甲酰氧基胺与降冰片二烯通过交叉偶联制备4-氨基吲哚的反应[59],并在PCM(Tol)/B3LYP-D3 /6-31G(d,p)-LANL2DZ(Pd,P,Cs,I)水平上对反应机理进行了DFT研究。结果显示:当底物R38中的R为叔丁氧羰基时,Cs2CO3以内辅助形式协助N—H和C—H活化的Gibbs自由能垒分别为16.9(TS38)和24.5 kcal·mol-1 (TS39, 图式22 a、b),说明反应更倾向于发生N—H活化,P24为主产物。当底物R38中的R为叔丁基时,Cs2CO3以内辅助形式协助N—H和C—H活化的Gibbs自由能垒分别为26.2(TS41)和25.3 kcal·mol-1 (TS42, 图式22 a、c),说明反应更倾向于发生C—H活化,P25为主产物。上述理论计算结果与实验结论一致,同时还表明Cs2CO3辅助N—H活化过程的能垒会受底物取代基的影响,这对于筛选和拓展C-N偶联反应的底物类型具有重要的参考价值。
5 Cs2CO3辅助钯催化B—H官能团化
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
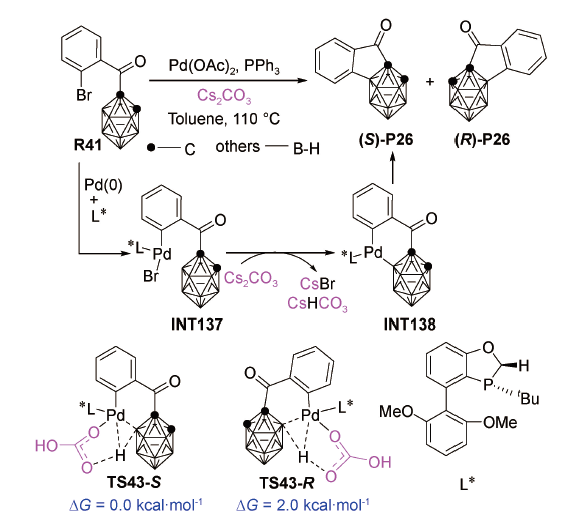
结果显示:Cs2CO3以内辅助形式协助B—H活化,形成(S)-P26产物需要跨越的过渡态TS43-S比形成(R)-P26产物需要跨越的过渡态TS43-R的相对Gibbs自由能低2.0 kcal·mol-1,表明反应更倾向于生成(S)-P26产物。他们的理论计算工作不仅很好再现了实验结果,还印证了Cs2CO3辅助钯催化B—H活化的可行性。2019年,本课题组在PCM(Tol)/LC-ωPBE/ DGDZVP-SDD(Cs)水平上对Cs2CO3辅助镍催化B—H活化反应进行了DFT研究[112],发现Cs2CO3(用K2CO3代替计算)以外辅助形式协助B—H活化的Gibbs自由能垒仅为23.1 kcal·mol-1,这表明Cs2CO3不仅能辅助钯催化B—H活化,在辅助其他过渡金属催化的B—H活化反应中也有着广阔的应用前景。
6 结论与展望
近十年,国内外化学工作者在Cs2CO3辅助钯催化X—H (X=C、O、N、B)官能团化领域开展了大量的理论研究工作,为人们深入认识Cs2CO3在辅助钯催化X—H (X=C、O、N、B)键活化反应中所起的作用及其微观反应机制等提供了极大的帮助。基于使用B3LYP、M06L和B3LYP-D3等方法的计算结果,Cs2CO3通常以直接或间接两种方式辅助钯催化X—H (X=C、O、N、B)键活化反应,其中直接辅助又分为内碱辅助型和外碱辅助型两种类型。在钯催化X—H (X=C、O、N、B)键的活化反应中,Cs2CO3多以直接方式辅助反应,其中內碱辅助型居多。大多数反应中,Cs2CO3均是通过直接参与反应,使相应中间体和过渡态的相对Gibbs自由能不同程度降低,从而有效降低反应的活化能垒,使反应易于进行的;自身则以CsX (X=Cl、Br、I、…)、CsHCO3、或CsX·CsHCO3团簇等形式从反应中脱去。上述反应机理的明确,为实验研究者有针对性地改变实验条件以降低决速步能垒、进而促进反应的快速高效发生提供了重要的理论指导。此外,关于不同碱添加剂对反应能垒的影响、以及对反应选择性的计算验证结果,将有利于实验上筛选出最优的碱添加剂,为后续实验在合理评估X—H (X=C、N、O、B)官能团化反应的主副产物比例基础上、采取合理措施得到预期产物奠定了基础。
在科研工作者的不懈努力下,Cs2CO3辅助钯催化X—H (X=C、O、N、B)官能团化反应取得了许多令人瞩目的成就,人们对Cs2CO3的辅助活化方式及作用机制等的认知也日趋成熟,但目前仍存在一些需要解决的问题:(1) 在实验研究方面,Cs2CO3辅助钯催化N—H和B-H官能团化反应的类型还有待拓展,Cs2CO3辅助钯催化的其他X—H官能团化反应如Si-H官能团化反应等,仍待进一步开发尝试。(2) 在理论研究方面,Cs2CO3辅助钯催化O—H和N—H官能团化反应机制的理论研究还有待深入,Cs2CO3辅助钯催化B-H官能团化反应的微观机制、区域选择性、化学选择性等问题仍缺乏系统的理论研究与探讨。
为此,继续拓展Cs2CO3辅助钯催化X—H (X=O、N、B、Si、…)官能团化的反应类型并开展DFT理论研究,在深入开展微观机理和反应选择性理论探究的基础上,设计开发更加经济高效的Cs2CO3辅助钯催化X—H官能团化反应体系并应用于C—X键的构建,将会是该领域未来的潜在发展方向。