



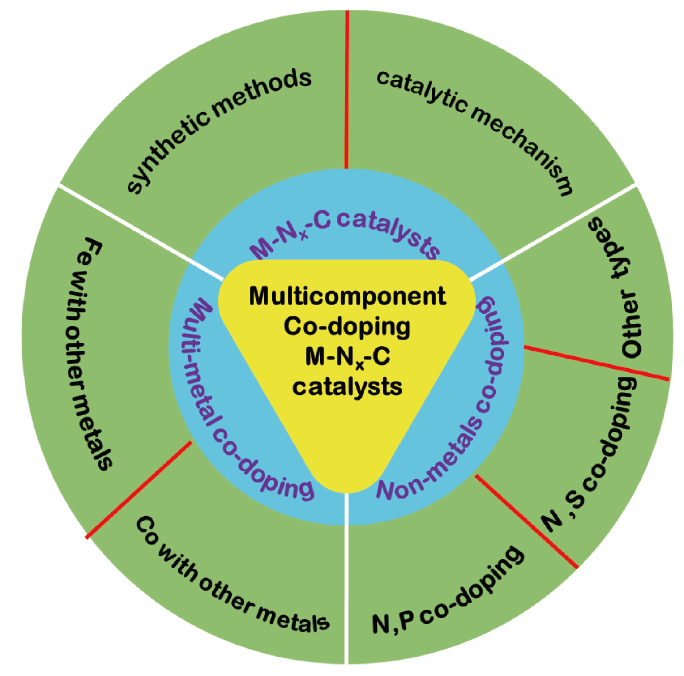
Contents
1 Introduction
2 M-Nx-C catalysts
2.1 The synthetic methods of M-Nx-C
2.2 The catalytic mechanism of M-Nx-C
3 Multi-metal elements co-doping M-Nx-C
3.1 Fe with other metals co-doping
3.2 Co with other metals co-doping
4 Non-metals elements co-doping M-Nx-C catalysts
4.1 N, S co-doping
4.2 N, P co-doping
4.3 Other co-doping types
5 Conclusion and outlook
1 引言
自工业革命以来,由于人类社会发展对化石燃料的过度依赖,致使地球上二氧化碳等温室气体含量骤增,由此带来的气候问题对自然和人类社会的可持续发展造成了严重威胁。我国政府更是做出“中国二氧化碳排放力争于2030年前达到峰值,努力争取2060年前实现碳中和”的庄严承诺。
在已报道的非铂催化剂中,具有类卟啉结构的过渡金属掺杂碳基催化剂(M-Nx-C)因其原料廉价易得和与铂可媲美的氧还原活性(尤其在旋转圆盘电极测试中),成为目前最为重要的一类非贵金属催化剂[7⇓⇓⇓⇓~12]。然而,由于M-Nx-C催化剂本征活性/活性位密度低、易腐蚀等问题,在单电池测试中,性能与商业Pt/C仍然存在一定差距。近年来,广大科研人员在提升M-Nx-C催化剂的活性和稳定性方面开展了大量研究,并取得了诸多进展。基于此,本文综述了近年来在M-Nx-C催化剂领域通过调控活性位点的电子结构与空间构型提升M-Nx-C催化剂的氧还原活性和稳定性的一些进展,具体包括金属元素掺杂、非金属元素掺杂等研究,这些工作将对此领域的进一步发展具有重要意义。
2 M-Nx-C单原子催化剂
2.1 M-Nx-C单原子催化剂及制备方法
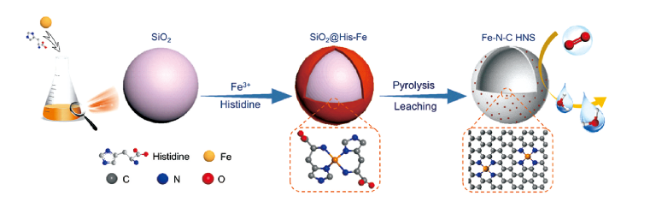
硬模板法。通常将前驱体填充到具有刚性骨架的硬模板中,控制时间让前驱体进行化学或电化学反应,然后进行高温碳化和酸/碱处理除去模板,最后得到形状有序的多孔纳米材料[19]。目前,二氧化硅纳米球[20]、金属氧化物[21]等是较为常用的硬模板。Chen等[22]利用SiO2纳米球做硬模板。实验过程中,首先对SiO2纳米球进行表面改性,使之带有负电荷,Fe3+通过静电作用吸附在纳米球表面。随后,组氨酸分子通过配位作用与Fe3+发生强结合,从而包覆在二氧化硅纳米球表面。再经过高温热解,退火,HF酸洗去除SiO2硬模板等一系列处理后,形成了具有中空形貌的Fe-N-C纳米碳球催化剂(合成示意图如图1 )。Chen等[23]则利用价格低廉的乳清粉作为碳源,氧化硅球和氯化锌粉末用作两种硬模板材料。制备过程中,需要将一定量的乳清粉溶解在适量丙酮中,再加入不同比例的ZnCl2和SiO2。将混合物搅拌均匀后,然后将其加热并在搅拌下干燥除去溶剂。磨成细粉的固体材料放入氧化铝舟中经过高温热解后,将得到的碳化材料首先在KOH溶液中回流以除去SiO2。再使用HCl溶液和大量去离子水洗涤以去除Zn残留物。实验通过调整两种硬模板的比例,实现了碳材料分级孔道结构的调控和优化。
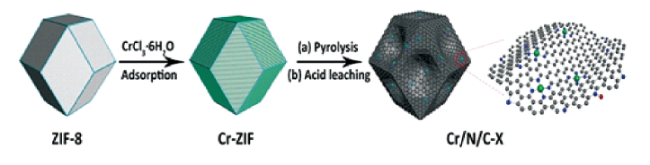
值得注意的是,硬模板法制备M-Nx-C催化剂时,常需要一些强酸/强碱去除模板,这不但导致制备流程冗长繁杂,而且易造成纳米结构的坍塌,难以很好地复制模板剂的多孔结构[24],而软模板法不但可以提供有序的立体结构,而且可以避免去除模板的繁琐步骤。目前,常用的软模板主要包括表面活性剂和聚合物,如表面活性剂P123、F127等嵌段共聚物[25,26]。在合成过程中,首先通过分子间或分子内的相互作用(氢键、静电等)使分子间形成具有某些特定结构特征的聚集体或胶束,随后以这些聚集体或胶束作为模板,允许无机物质通过各种合成方法沉积在其表面或内部,产生具有不同形态和多孔结构的颗粒[27]。例如,Lee等[28]报道了一种简单的软模板法,通过在嵌段共聚物自组装过程中直接掺入Fe-CNAR前驱体(Fe2+与1,10-菲咯啉的配合物),合成了具有FeNx/C活性中心的有序介孔碳材料。制备过程中,他们先将F127溶解在乙醇和HCl的共溶剂中,得到聚合物溶液,再将FeCl2·4H2O和1,10-菲咯啉溶于乙醇和HCl的溶剂中混合得到铁络合物溶液。最后将甲阶酚醛树脂、原硅酸四乙酯和铁络合物溶液添加到聚合物溶液中。搅拌1 h后,将溶液倒入盘中。经过50℃蒸发和100℃的烤干后,将得到的聚合物纳米复合材料经高温处理、酸洗和二次热解后即可得到制备的碳基催化剂。实验发现形成稳定的微观胶束结构是软模板法的关键,然而当前能用作表面活性剂的化合物相对较少,且在制备过程中,软模板法难以有效控制产物的形貌和尺寸。近来,因为MOFs具有规整的三维结构、高的比表面积以及高度有序的孔道[29⇓~31],利用MOF自模板合成M-Nx-C催化剂的方法也受到了很多关注。例如,Xing等[32]利用ZIF-8材料的有序孔道结构,CrCl3·6H2O为铬源,热解后合成了具有Cr-N4构型的单原子铬催化剂。合成示意图如图2 。制备过程中,将CrCl3·6H2O和ZIF-8分散到乙醇溶液中,在超声过程中铬离子被吸附到ZIF-8的微孔结构中,热解过程中Cr被碳载体中的CNx结构捕获/固定形成了CrN4结构。
尽管单原子催化剂的制备方法已被广泛研究,但实现单原子催化剂小批量(克级)甚至大批量(公斤级)的稳定工业化生产仍然是一个巨大挑战。最近,气相沉积法为解决单原子催化剂的大规模工业化制备提供了新思路[33]。采用CVD法制备M-Nx-C催化剂大致可分为两类:1)多孔纳米材料捕获易挥发的的金属化合物,即在气流作用下,含金属原子的易挥发化合物(Fe[34]、Mn[35]、Cu[36]等)通过气相迁移到多孔纳米材料表面被吸附;2)以掺杂Fe等过渡金属的金属氧化物(CaO、ZnO等)为模板,前驱体作为氮源和碳源沉积到模板上,在气相沉积过程中,可控碳化和原位杂原子掺杂有利于锚定单个金属原子以实现高的金属负载和活性位点M-Nx的有效暴露[33,37]。Wu等[36]利用NH3和泡沫铜表面铜原子的配位效应,将块体金属直接转化为单原子催化剂。在制备过程中,将铜泡沫和ZIF-8置于瓷舟中。首先,ZIF-8在高温的氩气氛围下热解,形成缺陷位点的热解C-ZIF-8。在氨气氛围下,氨气分子将表面的铜原子从铜泡沫中分离出来,形成基于强路易斯酸碱作用的挥发性Cu(NH3)x分子。然后,Cu(NH3)x被富氮碳载体捕获,高温热解后产生Cu-SAs/N-C单原子催化剂。Wu等[38]通过溶胶-凝胶法将Fe(NO3)3·9H2O与Zn(Ac)2·2H2O混合制备Fe-掺杂的ZnO基底。然后,将2-甲基咪唑和Fe-ZnO纳米片分别放在两个温度区中。蒸发的气态2-MeIm随氩气流动,随后沉积在Fe-ZnO纳米片上,得到了单原子Fe高度分散的多孔碳催化剂。
2.2 M-Nx-C单原子催化剂的催化原理
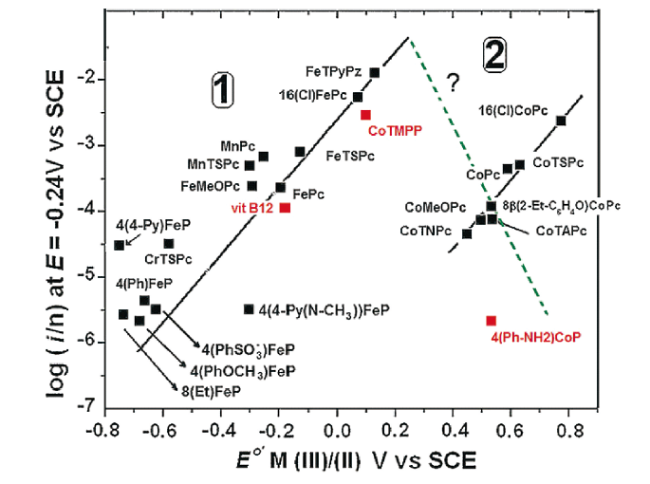
氧还原反应是一个多电子、多质子、多步骤的复杂反应,在不同电解液条件下,ORR的还原路径和还原产物也不同。根据不同反应路径,氧还原反应可分为四电子还原和两电子还原两种路径[39]。两电子路径常涉及到过氧化氢的生成,在芬顿反应下,过氧化氢会被还原产生自由基,而这些自由基会攻击质子交换膜,继而导致膜的降解,造成电池性能急剧下降。因此,理性设计催化剂催化氧还原按照四电子路径进行,避免过氧化氢的产生,可显著提升催化剂稳定性。而研究催化剂活性位点的催化机制是理性设计催化剂的关键。目前普遍认为,M-Nx-C催化剂的催化位点包括非金属部分和金属部分,相比于非金属部分(C-Nx),含金属部分MNx被认为是M-Nx-C催化剂高活性的来源。典型的金属活性位点,由非贵金属原子M(以Fe、Co、Ni、Cu、Zn等元素为主[40,41])与其配位的氮原子或其他原子组成。而MNx位点的配位结构可能因不同的合成方法和前驱体而异。Shui等[42]通过控制热解温度,制备出了含有相同活性位点密度,但是N原子配位数不同(配位数从1到5)的Fe-N-C催化剂。通过电化学表征和理论计算证明,FeN4具有最好的ORR活性和优异的PEMFC性能。此外,催化剂的氧还原活性与MNx结构中金属元素的种类有着密切的关系。Nørskov等[43]、Zagal等[44]、Qiao等[45]分别通过DFT计算和一系列实验建立了不同MN4结构的氧还原活性火山图,如图3 。研究发现,O2的吸附和OOH-的解离在碳基催化剂催化氧还原反应的过程中起着至关重要的作用。Peng等[46]利用三聚氰胺、苯胺和一系列过渡金属的氯化盐,制备了一系列氮和过渡金属共掺杂的催化剂(过渡金属=Mn、Fe、Co、Ni、Cu),通过实验证明了不同金属中心M—N—C催化剂的活性为:Fe>Co>Cu>Mn>Ni。显然,适中的氧吸附促进O=O解离是获得高活性的关键。另一方面,研究人员一直试图揭示ORR反应中的活性位点和反应中间体的动态转变过程。
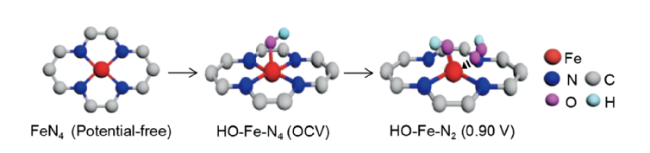
Liu等[47]运用基于同步辐射的表征技术,原位探测到了具备不饱和配位的铁活性结构OH-Fe-N2在反应过程中的动态变化。如图4 所示,开路电压下,活性中心发生羟基基团的预吸附,形成HO-Fe-N4配位结构。0.90 V的氧还原电位施加时,由于OH的牵引作用,较长的两配位Fe-N路径发生断裂,形成配位不饱和的HO-Fe-N2催化活性结构。同时,活性位点上发生关键反应中间体(*OOH)的动态吸附及脱附作用。HO-Fe-N2活性结构在催化过程中保持稳定,保证了氧还原反应的高效进行。
3 多种金属共掺杂的M-Nx-C单原子催化剂
近几年的研究结果显示,单一过渡金属类型的金属-氮-碳催化剂在碱性介质中具有可与商业铂碳相媲美的ORR活性[50],然而,在酸性介质中的性能仍然有一定的差距。可能的原因是这类催化剂的金属负载量低,且催化过程中孤立金属原子易发生聚集和溶出,导致催化剂活性和稳定性不足[51,52]。最近,有研究报道向单一过渡金属M-Nx-C中引入第二组分金属元素,改变单金属活性位点的几何构型和周围的化学环境可提高M-Nx-C活性和稳定性[53]。在诸多过渡金属(如,Fe、Co、Cu,Ni、Mn等)掺杂碳基催化剂中,以Fe、Co为中心的M-Nx-C催化剂因具有最佳氧还原活性而被广泛研究[44,54,55]。现总结如下。
3.1 铁与其他金属共掺杂
Sun等[56]合成了一种具有FeNi-N6活性位点的单原子电催化剂。ICP-OES检测到该催化剂中的Fe和Ni的百分比分别为1.45wt%和1.47wt%,金属总含量接近3wt%。镍金属位点的引入提供了大量的催化活性位点,同时使该催化剂在ORR过程中的H2O2产率仅为1%~4%。酸性条件下,FeNi-N6展现出了优异的耐久性,5000圈循环稳定性测试后半波电位衰减仅为12 mV。Chen等[57]通过金属-有机骨架(ZIF-8)一步热解制备了具有两种金属M-Nx活性位点的电催化剂Fe,Mn-N/C。LSV测试结果显示含有Fe-Nx和Mn-Nx位点的催化剂比只有Fe-Nx活性位点的催化剂具有更优异的ORR活性。Fe和Mn离子之间的协同作用有效降低了ORR过程的能垒,通过第一性原理计算得出,与Fe-N/C相比,在ORR过程中,Fe/Mn-N/C在从O*到OH*的质子化过程中所需的能量更少。该催化剂表现出卓越的ORR性能,半波电位为0.904 V,电流密度为33.33 mA·cm2, 是 20% Pt/C (6.76 mA·cm2)的4.9 倍。
Chen等[58]通过混合铁铜金属盐、PVP和纳米CaCO3颗粒,简单地制备了前驱体,在经过一系列处理后,得到了具有FeN4和CuN4单原子共存的高效催化剂。电化学测试表明,具有双金属位点的Fe/Cu SA活性明显好于单位点的Fe SA或Cu SA,表明了Cu位点对Fe位点ORR活性的促进作用。从文献中的电子局域函数(ELF)图像可以看出,在Fe/Cu SA结构中,铁原子的电子密度明显增加。Bader电荷计算表明,引入Cu后,Fe的电荷转移数从-0.96下降到-0.88 e。这有力地证明了相邻位点在调节铁原子电子结构中的关键作用,即阻止铁原子的电荷密度下降,从而提高铁原子周围的电子密度。较高的电子密度促进了O2分子的吸附和活化。
Shui等[59]利用密度泛函理论研究了Zn对Fe-N-C催化剂电催化性能的影响,计算结果表明,Fe-Zn-Nx结构在热力学上是稳定的,第二金属锌原子通过修饰Fe-N-C的电子结构,削弱了Fe-O原子之间的结合强度,降低了ORR活化能垒,从而提高了Fe-N-C的催化活性。
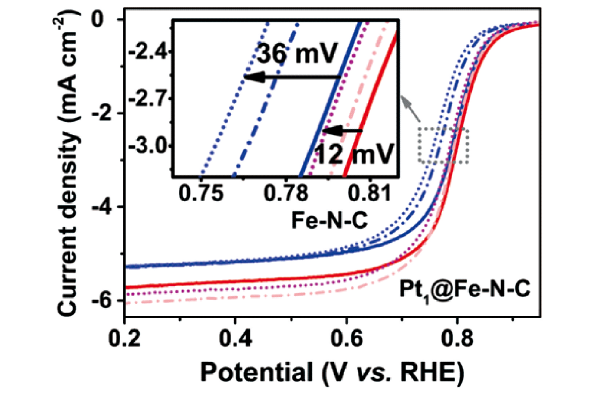
此外,Shui等[60]也利用铂原子对Fe-N-C催化剂进行原子尺度的修饰,他们将单原子Pt通过桥氧分子嫁接到Fe-N4活性中心,得到新活性位点Pt1-O2-Fe-N4。催化剂在酸性介质中表现出略微增强的ORR活性,且在全电池中表现出显著增强的稳定性。在0.3~1.1 V之间,对Pt1@Fe-N-C和Fe-N-C进行了加速电压循环测试,如图5 所示,Pt1@Fe-N-C的E1/2在10 000个循环后仅衰减了12 mV,比Fe-N-C的36 mV的衰减量要小得多。这些结果表明,将Pt1O2接枝到Fe1N4分子上是一个有效的策略来提高Fe-N-C电催化剂的耐久性。这种稳定性的增强得益于Pt1-O2-结构保护Fe位点免受双氧水破坏。进一步证明了接枝原子Pt与原始活性中心Fe之间的协同作用。这个研究也通过单原子与单原子嫁接技术为提高和拓展碳基金属催化剂性能提供了一个新的思路。
3.2 钴与其他金属共掺杂
相对于Fe-N-C催化剂而言,Co-N-C催化剂拥有较低的芬顿反应活性。最近有研究发现,通过构筑Fe和Co双金属位点可以进一步增强金属中心的催化活性。Xing等[61]利用Fe-ZnCo-ZIF-8前驱体制备了Fe和Co都是以原子形式分散且具有Fe-Co双原子活性位点的碳基催化剂。在此催化剂的活性位点上,能够自发吸电子的OH配体被引入,降低Fe位点的d带中心并与Fe-Co双原子活性位点形成三角形Fe-Co-OH配位。在Fe-Co-OH活性位点上,ORR中间体的结合程度减弱,O—O键断裂加速,ORR活性增强。Zhang等[62]也报道了使用甲酰胺作为前驱体大规模生产Fe-Co-NC双金属原子催化剂的一般路线。
Sun等[63]通过热解前驱体制备了一种具有Zn/Co双金属位点的Zn/Co-N-C催化剂,该催化剂在酸性和碱性条件下都表现出了优异的催化活性,如图6 所示,在0.1 M HClO4中,半波电位高达0.796 V vs RHE。将该催化剂用于H2-O2 PEMFC时,其峰值功率密度达到705 mW·cm-2。

Hu等[64]通过在碳纳米管上热解钴镍金属盐和三聚氰胺树脂得到了钴镍负载的氮掺杂碳纳米管(CoNi-NCNT)。通过旋转环盘电极测试技术表明氧还原反应在Co/Ni-NCNT催化剂上以接近理想的四电子转移路径进行,双氧水产率低于5%,性能远远领先于Co-NCNT。他们提出Ni的引入能够与Co-N-C结构协调形成Co/Ni-N-C结构,将很大程度上提高电子转移数和活性中心稳定性。
Amal等[65]制备了铜钴二元催化剂,研究发现钴单原子位点附近铜单原子位点的存在使得钴单原子活性位点处的电子结构发生了改变,使其有利于氧还原反应的发生,从而大幅提升了表观反应活性。此外,钴铜二元催化剂相比钴单原子催化剂有着更低的过氧化氢选择性,有效提升了催化剂的反应稳定性、活性,使其比商用20% Pt/C有着更好的活性与稳定性。理论及实验表明,富电子的钴单原子在反应过程中是氧还原的活性位点,而铜单原子则主要充当了过氧化氢抑制剂的作用。
4 非金属元素掺杂M-Nx-C单原子催化剂
除金属元素掺杂以外,通过掺杂非金属元素(S、P和B等)打破平面M-N4结构的对称性也可有效调控它们的电子结构并优化中间产物的吸附强度以提高ORR活性。
4.1 N、S共掺杂
在氮、硫共掺杂的碳基催化剂中,具有比氮更大原子半径和更低电负性(χ=2.58)的硫可以诱导电子自旋重新分布和极化构型转变,增强碳基催化剂的ORR活性。
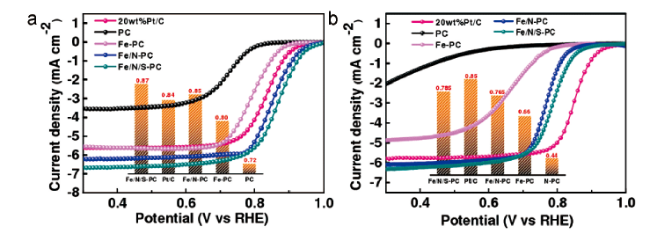
Chen等[66]通过Fe穆斯堡尔谱和电子顺磁共振光谱证实S掺杂可以诱导Fe自旋极化构型的转变,S掺杂后较低的自旋状态促进了OH*的解吸过程,同时提高了催化剂的选择性,促使整个ORR过程为四电子转移路径。本课题组[67]利用双溶剂扩散热解法制备了一种Fe、N、S共掺杂的UIO-66—NH2衍生碳催化剂(Fe/N/S-PC)。硫掺杂提升了氮活性位点的数量,形成的噻吩结构改变了碳骨架的电子结构,除此之外,与无硫掺杂的催化剂相比,该衍生碳材料的分层多孔结构使其具有更高的比表面积和介孔率,进一步促进了氧气和电解质的传输,如图7 所示,Fe/N/S-PC在酸性和碱性下均展现出了优于商业 Pt/C的活性,且具有更大的电流密度。Fe/N/S-PC的优异ORR性能归因于其高的介孔孔隙率、高活性的活性位点和大的比表面积。
硫原子掺杂到M-N4活性位点周围的不同位置会使催化剂的电子结构和ORR步骤的吸附自由能存在差异,从而导致其对碳基催化剂活性提升的程度不同。Sun等[68]基于甲酰胺液相合成体系,通过设计分子尺度的反应,精准实现Fe-N4外围不同位置S元素的精确掺杂(如图8 a所示)。DFT模拟结果显示Fe-NS2-C中Fe的d带隙远窄于其他Fe-NS-C中的Fe原子,这归因于相邻的S掺杂提高了Fe-N4部分的局部电子电导率。除此之外,结合电镜技术和同步辐射技术,他们发现催化剂活性位点的原子结构为Fe(N3)(N-C-S)构型,Fe-N4近邻硫掺杂的Fe-NS-C材料表现出优异的氧气还原催化活性和稳定性。如图8 b所示,Fe-NS-C的起始电位约为1.09 V,分别超过Pt/C和 Fe-N-C 的起始电位90和80 mV。
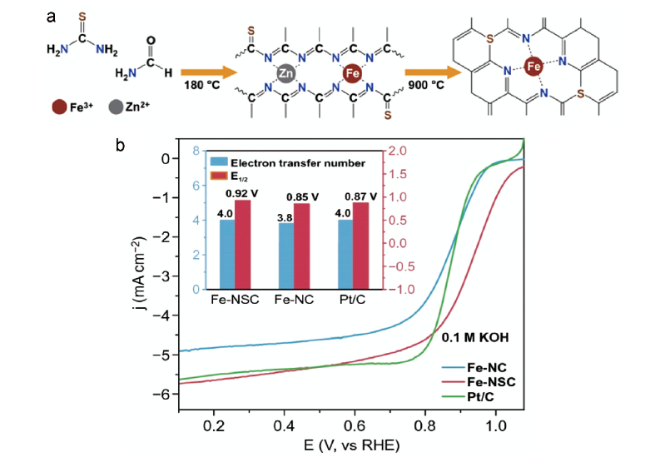
图8 (a) Fe-NSC的合成示意图;(b) 0.1 mol/L KOH 中的LSV 曲线(转速: 1600 r/min);插图为相应的半波电位和转移电子数[68]Fig. 8 (a) Schematic illustration of the synthesis of FeNSC. (b) ORR LSV curves for different catalysts in 0.1 mol/L KOH solution (rotation rate: 1600 r/min). Inset shows corresponding half-wave potentials and the number of transferred electrons[68] |
Zhu等[72]通过一步热解包覆FeCl3的紫菜前驱体,合成了N、S共掺杂的Fe-N-C材料。经过同步辐射对Fe原子的配位环境进一步分析之后,发现Fe-N与Fe-S局部配位键的比率接近3∶1,推测催化剂活性位点为Fe-N3S1。电化学测试结果表明,与不掺S元素的催化剂相比,硫掺杂的催化剂表现出了更好的ORR活性和四电子选择性。ORR活性的提升归因于硫掺杂对Fe-Nx结构电荷和自旋分布的优化。
除单金属位点催化剂外,杂原子掺杂也常常应用于具有双金属位点的碳基催化剂中。Zhao等[51]以壳聚糖为碳源、二乙基二硫代氨基甲酸钠为硫源、醋酸钴为钴源、氯化锌为锌源,通过简便的同步配位热解方法合成了硫掺杂改性的Zn,Co-Nx-C-Sy双金属催化剂。实验结果证明,这种活性位点在ORR催化中具有两个优势:(1)Zn-Co双金属位点增强了与O2的结合,促进了O—O键活化,并降低O—O键在*OOH+e-→*O+OH-步骤中的解离势垒;(2)S掺杂可以在Zn、Co活性中心周围调控电荷,并通过降低*O2+e-+H2O→*OOH+OH-步骤的自由能,来加强与含氧物质的相互作用。电化学测试表明,制备的Zn,Co-Nx-C-Sy表现出优异的电催化性能,半波电位比商用Pt/C高67 mV(0.893 V vs 0.826 V),计时电流测试显示样品具有优异的稳定性(20 000 s测试后损失电流约4.4%)。
4.2 N、P共掺杂
相比于氮原子而言,磷原子具有更大的原子半径及低的电负性,P取代部分N原子与金属配位时,P与N的物理化学性质差异引起的不对称N/P耦合会优化活性中心的电荷分布和电负性,亦可提升催化剂的性能。
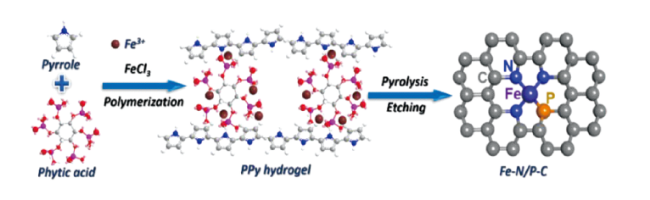
Chen等[73]开发出一种氮和磷双配位单原子铁碳基催化剂。如图9 所示,通过吡咯的聚合反应形成聚吡咯水凝胶,并引入植酸和FeCl3作为磷源和铁源。随后将水凝胶在氮气中热解,并使用硫酸刻蚀得到氮、磷双配位的铁活性位点复合碳纳米片。利用多种表征和模拟计算的手段,证明了催化剂中的Fe原子与三个氮原子和一个磷原子配位,形成方形平面构型。通过实验和理论计算表明,N、P双配位的铁位点有利于氧中间体的吸附/解吸过程,可以加快氧还原反应动力学,并实现高催化活性。
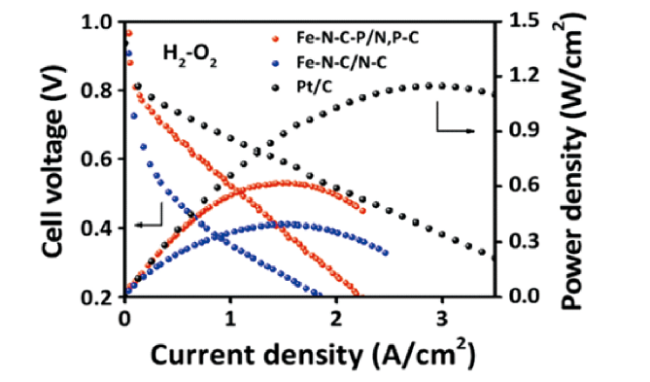
Zhang等[74]研究了远离的磷原子通过远程电子的离域作用提高边缘型FeN4催化活性的原理。通过DFT计算研究了边缘型FeN4P2的局部电子及分布,发现相较于平面内的FeN4P2,边缘型的FeN4P2结构的缺陷调节作用更有利于四电子ORR途径。实际的PEMFC性能测试也表明催化剂具有良好的活性和稳定性,其在H2/O2和80℃下,电池表现出0.98 V的高开路电压和0.625 W/cm2的最大功率密度,如图10 所示。
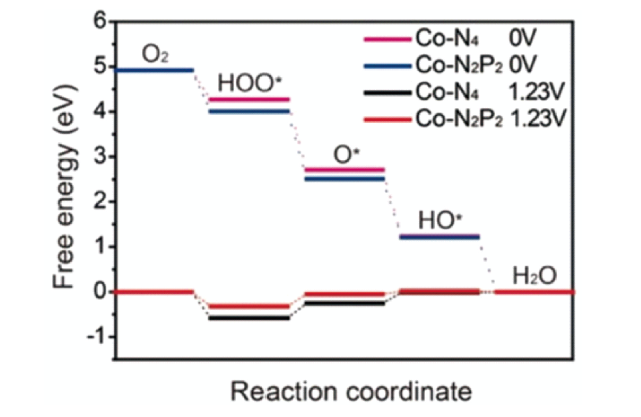
Zhang等[75]开发了一种新型交联聚磷腈球,碳化后得到N/P与Co配位形成Co-N2P2位点的介孔碳纳米球。与传统的热解之前格栅化引入杂原子的方法不同,交联共聚体提供的N、P等杂原子可以直接与金属配位得到均匀的活性位点,使催化剂中Co含量高达10.12%。如图11 ,DFT计算结果显示,与Co-N4位点相比,O2分子在Co-N2P2上有更低的吸附自由能,表明Co-N2P2可以更有效地结合O2来催化ORR。
Li等[76]使用磷掺杂的金属有机骨架(MOF)作为多孔载体,通过多步合成制备具有Co1-PNC和Ni1-PNC平面构型的单原子催化剂,得益于N/P配位的 Co/Ni 位点对氧中间体结合相互作用的适度降低,该催化剂在碱性溶液中表现出优异的ORR催化活性。在O2饱和的0.1 mol/L KOH溶液中进行的电化学测试表明,Co1-PNC/Ni1-PNC显示了最优的ORR起始电位和半波电位(Eonset=1.00 V, E1/2= 0.88 V),优于实验中的其他样品性能。此外,Co1-PNC/Ni1-PNC的Tafel斜率值(63 mV/dec)低于Pt/C(87 mV/dec)和Co1-NC/Ni1-NC(93 mV/dec),较低的Tafel斜率暗示了出色的ORR动力学。
4.3 其他多种非金属元素共掺杂
除了上面提到的N/S、N/P的共掺杂方式外,近年来,N/Cl、N/O、N/B等共掺杂方式也被探究应用于M-Nx-C催化剂中。
Li等[77]通过热迁移法制备了硫、氯元素共掺杂的FeCl1N4/CNS催化剂。在活性位点中,Cl、N原子位于金属位点的第一配位壳,S原子则距离金属活性位点较远。所获得的催化剂表现出非常优异的ORR活性。电化学测试表明它在0.1 mol/L KOH溶液中的半波电位为0.921 V,比商用Pt/C(0.842 V)高79 mV。这项工作通过实验和DFT证明,通过同时调节Fe活性位点与氯的近程相互作用和与硫的长程相互作用,可以成功实现ORR的高度改善。

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
5 结论与展望
本文系统地综述了近年来国内外在多种金属/非金属元素掺杂提升原子级别分散的M-Nx-C碳基催化剂性能方面的主要研究工作。显然,经广大研究者的不懈努力,原子级别分散的M-Nx-C催化剂的氧还原性能已获得诸多进展,展现出诱人的前景,然而要促进其在燃料电池膜电极中的实际应用仍存在诸多挑战。
第一,单原子活性中心面临着如何提高ORR选择性、降低过氧化氢产率的问题[30]。催化过程涉及到化学键的断裂和形成[81]。M-Nx-C催化剂因为活性位点的空间构型,氧气更倾向于以超氧态吸附在位点上,更易在后续的反应中产生过氧化氢。如果将活性位点的金属原子由单个原子增加到两个以上,形成M2-N-C甚至Mx-N-C结构,活性位点中存在两个以上的原子破坏O—O键,使得氧气以过氧态吸附到位点上,可以提高四电子反应选择性[30,82]。因此,未来M-Nx-C催化剂可能会朝着双原子、三原子或者原子簇型的M-Nx-C催化剂的方向发展。利用多核金属配位配合物或原子簇化合物中含有多个金属原子的特点,或许能较便利地合成Mx-N-C催化剂。
第三,难以做到对原子级活性中心结构的精确表征,解释单原子催化中心的理论仍不够完善。尽管目前已经有了透射电镜、球差电镜、同步辐射等仪器用于催化剂的表征,但是它们很难区分配位环境中的细微差别。尤其在非均相催化反应中,单原子催化剂的动态结构转变非常普遍。考虑到这一点,采用更多的时间分辨技术,如原位XAS、XPS、IR和Raman光谱等来跟踪反应中间态,检测活性位点在反应条件下的转变很重要[85]。因此,为了更好探索原子级结构的催化机理,开发和研究高精度的仪器是非常重要的。
表1 近年M-Nx-C催化剂电化学性能汇总表Table 1 Summary of electrochemical performance of M-Nx-C electrocatalysts |
| Catalysts type | structure | Carbon carrier | Acidic media | Alkaline media | ref | ||
|---|---|---|---|---|---|---|---|
| Eonesetvs RHE(V) | E1/2vs RHE(V) | Eonesetvs RHE(V) | E1/2vs RHE(V) | ||||
| Non-doped M-Nx-C | Fe-N-C | histidine | - | - | 1.046 | 0.87 | 22 |
| Fe-N-C | block co-polymer | - | - | 1.00 | 0.901 | 28 | |
| Co-N-C | the mixture of polystyrene, polyacrylonitrile | - | - | 0.95 | 0.86 | 88 | |
| Co-N-C | Zn/Co-ZIF | - | - | 0.982 | 0.881 | 30 | |
| Cr-N-C | ZIF-8 | - | 0.773 | - | - | 32 | |
| Cu-N-C | ZIF-8 | 0.83 | - | 0.99 | 0.895 | 36 | |
| Fe-N-C | 2-MeIm | 0.963 | 0.835 | - | - | 38 | |
| Fe-N-C | ZIF-8 | - | 0.78 | - | 0.864 | 34 | |
| Mn-N-C | ZIF-8 | - | - | - | 0.90 | 35 | |
| Non-metal elements doped M-Nx-C | Fe-N/S-C | UIO-66—NH2 | - | 0.785 | 0.97 | 0.87 | 67 |
| Fe-N/S-C | formamide | - | 0.78 | 1.09 | 0.92 | 68 | |
| Fe-N/S-C | porphyra | - | - | 0.96 | 0.84 | 72 | |
| ZnCo-N/S-C | chitosan | - | - | 1.07 | 0.893 | 51 | |
| FeN3P1 | - | 0.89 | 0.72 | 0.941 | 0.867 | 73 | |
| FeN4P2 | - | 0.80 | 1.06 | 0.87 | 74 | ||
| Co-N2P2 | - | - | - | - | 0.878 | 75 | |
| FeCl1N4/CNS | - | - | - | - | 0.921 | 77 | |
| Fe-N/S/P-C | ZIF-8 | - | 0.791 | - | 0.912 | 69 | |
| Co-N/B-C | - | - | - | - | 0.83 | 79 | |
| Mn-N/O-C | Mn-BTC | - | - | - | 0.86 | 70 | |
| Multi-metal elements doped M-Nx-C | Fe/Ni-N-C | ZIF-8 | - | - | - | 0.02* | 56 |
| Fe/Mn-N-C | ZIF-8 | - | - | - | 0.904 | 57 | |
| Fe/Cu-N-C | PVP | - | - | 0.96 | 0.86 | 58 | |
| Pt1-O2-Fe-N4 | - | 0.93 | 0.80 | - | - | 60 | |
| FeNi-N-C | polydopamine | - | - | 0.95 | 0.82 | 87 | |
| Fe/Co-N-C | ZIF-8 | - | - | 0.995 | 0.920 | 89 | |
| Fe/Co-N-C | Co/Zn-ZIF | 1.02 | 0.86 | - | - | 61 | |
| Zn/Co-N-C | Zn/Co-ZIF | - | 0.796 | - | - | 63 | |
| Co/Ni-N-C | CoNi-NCNT | - | - | 0.88 | 0.81 | 64 | |
| Co/Cu-N-C | DCDA | - | - | 0.98 | 0.88 | 65 | |
| Fe/Ni-N-C | polystyrene spheres,ZIF-8 | - | 0.840 | - | 0.938 | 90 | |
* negative than 20% Pt/C |