




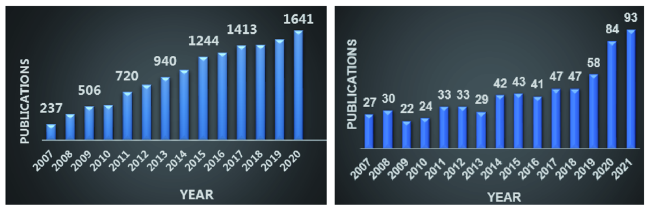
1 引言
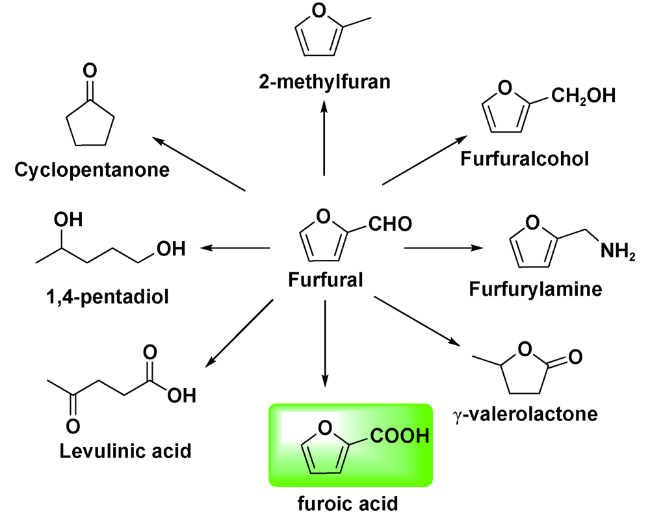
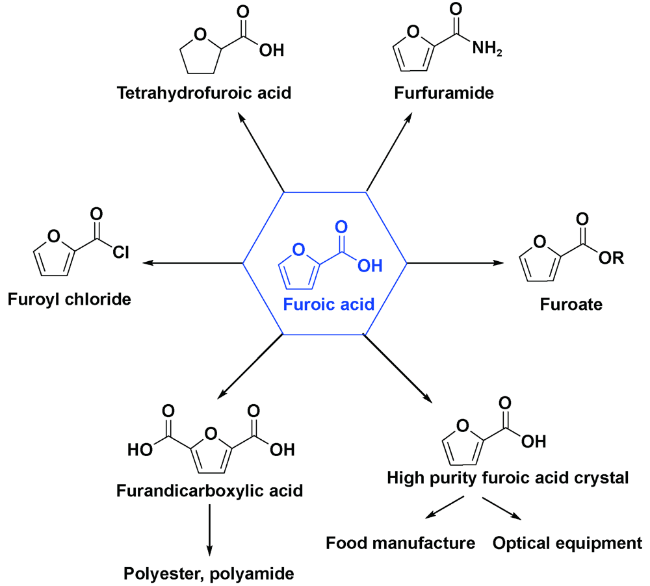
糠酸通过加氢反应可以合成重要的药物中间体四氢呋喃甲酸,进而制备多种已经具有临床应用的药物。糠酸经过酯化反应后可以得到糠酸酯,可以作为修饰剂和增香剂应用于食品、饮料和化妆品行业。糠酸的衍生物糠酰氯和糠酰胺可以用于制备多种精细化学品以及生物基聚合物。同时糠酸也可以通过固载二氧化碳合成新型生物基材料单体2,5-呋喃二甲酸(FDCA),进而合成生物基呋喃材料聚2,5-呋喃二甲酸乙二醇酯(PEF)[10]。PEF被认为是一种改进的包装材料,具有优异的阻水、阻氧及二氧化碳阻隔性,可以延长食品储存时间,降低加工温度。糠酸粗产物通过升华方式可以得到高纯糠酸晶体,可以直接用作食品的防腐剂、杀菌剂。同时由于糠酸晶体良好的结构特性,可作为非线性光学材料(NLO)应用到光学设备中[11]。
本综述整理了近年来通过催化氧化糠醛制备糠酸的研究情况,以均相/非均相催化体系进行了分类阐述,分析讨论了已报道的催化体系,重点突出了非均相催化剂在糠酸的工业化制备中的应用前景,展望了糠酸制备中值得重点关注和研发的方向。
2 催化氧化体系
我国的糠酸产业中主要存在生产规模小、数量少、生产工艺相对落后等问题,因此需要开发廉价环保的催化氧化方法,满足迫切的生产需求。传统的糠酸制备方法主要包括当量氧化剂氧化法、Cannizarro歧化法以及催化氧化法。其中,当量氧化剂如高锰酸钾、重铬酸钾法、次氯酸钠和过氧化钠等的使用存在一些局限性,如三废多、选择性低、氧化剂原子利用率低、氧化剂本身安全性等问题。Cannizarro歧化反应简单,一般在碱性条件下既可以实现两分子糠醛到一分子糠酸和一分子糠醇的转化,但是反应中生成当量的糠醇分离操作比较烦琐,且其结构不稳定,分离成本比较高[12]。目前糠酸的研究主要集中在催化氧化上。催化体系主要包括均相催化体系、非均相催化体系及生物氧化体系,下面将分别对三种类型的催化体系的反应情况进行阐述。
2.1 均相氧化体系
按照反应过程中所需的氧化剂的不同,将分别从以氧气、空气、过氧叔丁醇(t-BuOOH)及其他氧化剂为氧源来阐述均相催化体系催化糠醛到糠酸的反应情况,相关结果汇总在表1 中。
表1 均相催化体系催化转化糠醛到糠酸反应汇总Table 1 Summary of catalytic conversion of furfural to furoic acid by homogeneous catalytic system |
| Entry | Catalyst | Reaction conditions | Furoic Acid Yield/% | ref |
|---|---|---|---|---|
| 1 | [Cu(acac)2]/SIMes | H2O, 50 ℃, 1 atm O2, 1 eq NaOH, 12 h | 99 | 13 |
| 2 | triazolium-NHC | DABCO(50 mol%), O2, THF, r.t., 16 h | 96 | 15 |
| 3 | 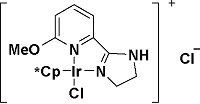 | NaOH (1 eq), H2O, air, 80 ℃, 12 h | 90 | 17 |
| 4 | 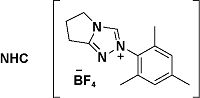 | MeCN, TBD(1.2 eq), air, r.t. | 86 | 17 |
| 5 | CuCl | 2 equiv of 70% t-BuOOH (in water) in MeCN, r.t., 3 h | 91 | 18 |
| 6 | CuBr2 | 2 equiv. t-BuOOH (water), MeCN, 1.5 h | 92 | 19 |
| 7 | Bi2O3 | 5 equiv. 70% t-BuOOH (water), EtOAc, 10 h | 91 | 20 |
| 8 | Mohr’s salt | DMSO, 5 eq. 70% t-BuOOH, 80 ℃, 3.5 h | 92 | 21 |
| 9 | I2 | NaOH (20 mol%), 4 equiv. t-BuOOH (water), H2O, 70 ℃, 10~16 h | 77 | 22 |
| 10 | PCC | Solvent-free, r.t. | 82 | 23 |
| 11 | Sodium chlorite | MeCN, 10 ℃-r.t., 3 h | 90 | 24 |
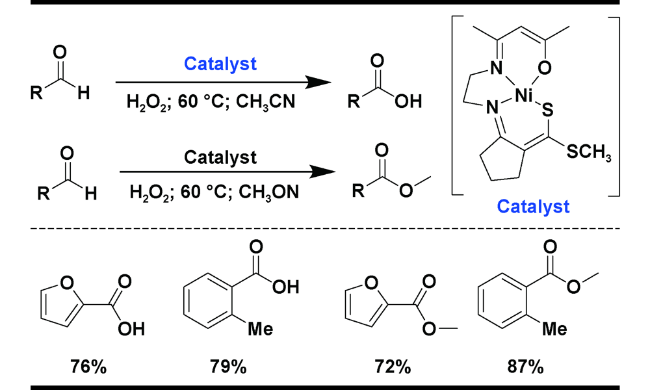
| 12 | 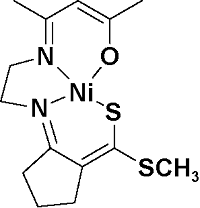 | H2O2; 60 ℃; CH3CN | 76 | 25 |
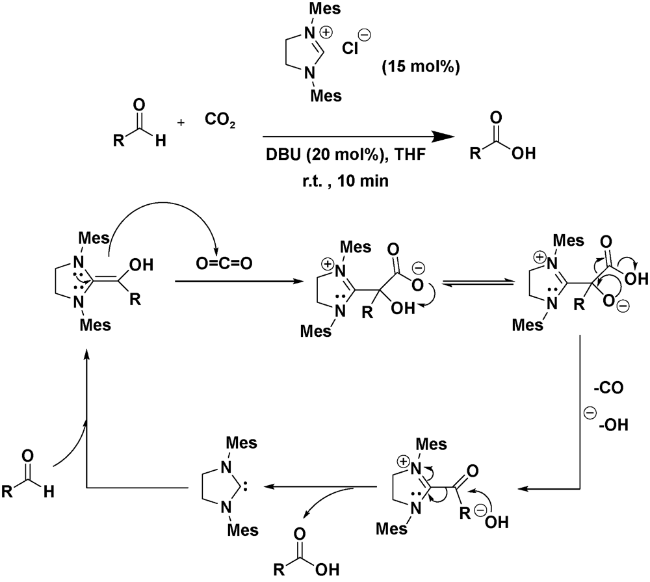
| 13 | 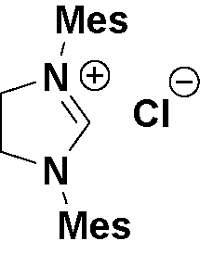 | CO2, DBU (20 mol%), THF, r.t., 10 min | 65 | 26 |
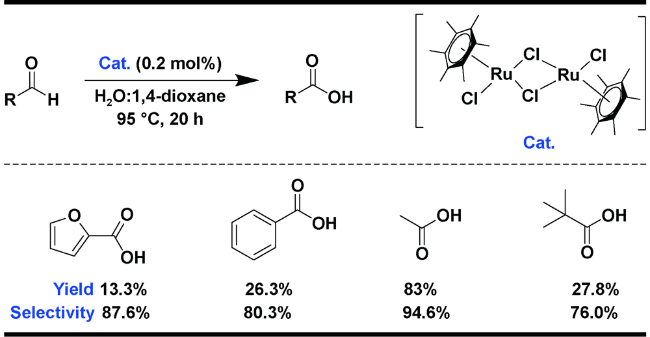
| 14 | 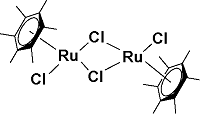 | H2O∶1,4-dioxane=50∶50, 95 ℃, 20 h | 13.3 | 27 |
2.1.1 氧气为氧化剂
在糠醛的氧化反应中,氧化剂的选择是至关重要的。空气或氧气作为绿色氧化剂,廉价且环保,反应中水是唯一的副产物。
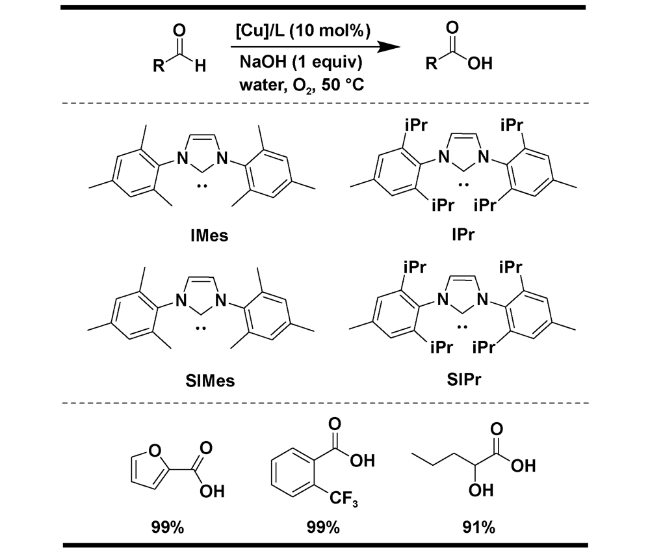
Li等[13]尝试了CuCl2、CuCl、CuBr2与多种配体(联吡啶、膦配体、IMes、IPr、SIMes、SIPr等)的组合,对醛的氧化进行了测试,最终发现更富电子的SIMes配体制备得到的催化剂对反应的氧化活性最好(图4 )。在氢氧化钠水溶液中,50 ℃反应12 h,使用[Cu(acac)2]/SIMes催化剂可以得到99%的糠酸分离收率。虽然[Cu(acac)2]/SIMes催化剂的制备过程相对简单,不过制备催化剂所需的原料1,3-双(2,4,6-三甲基苯基)- 4,5-二氢咪唑氯化物(SIMes·HCl)的价格比较昂贵,导致催化剂的成本较高(表1 ,序号1)。
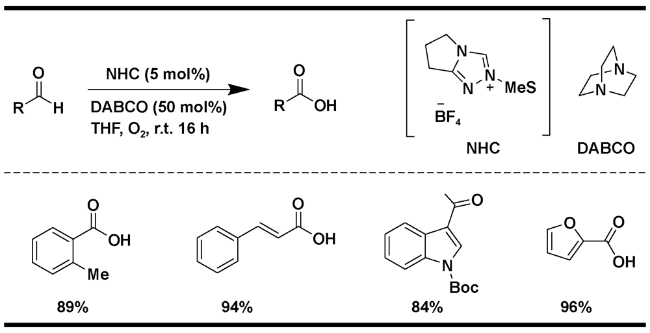
与[Cu(acac)2]/SIMes催化剂代表的金属配合物催化剂相比,无金属有机催化剂被视为一种有效的替代方式[14]。在所有基于有机分子的催化剂中,N-杂环卡宾(NHC)类催化剂已发展成醛氧化到羧酸最有前途的催化剂种类之一。Tiwari等[15]通过对多种N-杂环卡宾催化剂及碱添加剂的筛选,开发了一种高效的三唑钅翁-NHC催化剂,在碱DABCO存在下,四氢呋喃溶剂中,室温下氧气中反应16 h,可以将糠醛氧化得到糠酸,收率为96%(图5 )。该三唑钅翁-NHC催化体系对几类具有氧化挑战性的醛包括邻位取代芳醛、高度富含电子的芳醛和吲哚-3-甲醛等均具有较高的催化活性和羧酸产物选择性,该方案的反应条件温和,并且还可以用于克级规模合成(表1 ,序号2)。
2.1.2 空气为氧化剂
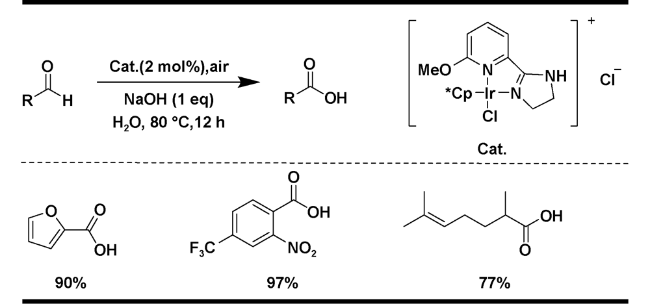
开发高效的催化体系,实现在水相体系中空气条件下的高效氧化是最理想经济的反应方式。在以空气为氧化剂的研究中,Tang等[16]报道了在氢氧化钠存在下,使用空气作为氧化剂,水作为溶剂,Ir络合物作为催化剂将醛直接氧化成酸,其中产物糠酸收率达到90%,产物不需要经过柱层析进行纯化即可以达到较高的纯度(图6 )。反应机理的研究表明,有氧氧化涉及高度还原的氢化铱,氢化铱可以捕获氧而得到氢氧化铱。有趣的是,该催化剂也用于在酸性条件下还原醛类化合物(表1 ,序号3)。
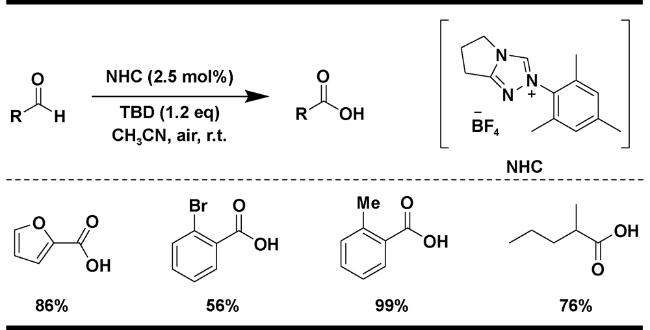
Studer等[17]开发了一种高效的NHC和Ru(Ⅱ)基氧化还原催化剂,在醇溶剂中,可以实现芳香醛和杂环醛的氧化酯化制备相应酯化产物(表1 ,序号4)。单独使用NHC作为催化剂,在乙腈溶剂,室温条件下,可以选择性地将糠醛空气氧化到糠酸,收率为86%(图7 )。
2.1.3 t-BuOOH为氧化剂
均相催化剂进行糠醛到糠酸的氧化,使用最多的氧化剂主要是过氧叔丁醇。Sekar等[18]报道了在室温条件下,过氧叔丁醇水溶液作为当量氧化剂,在乙腈溶剂中使用CuCl催化剂实现醛到相应羧酸的氧化(图8 ;表1 ,序号5)。首先使用DABCO-CuCl络合物/O2/TEMPO(2,2,6,6-四甲基哌啶氧化物)催化体系,但是该反应甚至没有提供痕量的相应羧酸。使用30%的H2O2代替氧气,在TEMPO催化剂存在下,羧酸收率有了一定的提升。当用无水t-BuOOH(5 mol/L在癸烷中)代替30%的H2O2时,所有原料在室温下仅6 h就被消耗掉,反应分离出的糠酸产率为91%,其他苯环类和脂肪族底物也可以得到较好的羧酸收率。他们意外地发现,该反应中真正的催化剂是不含配体的CuCl。单独的CuCl作为催化剂得到了相同的效果。该催化体系简单,催化剂廉价易得,进行优化避免使用无水t-BuOOH作为当量氧化剂或者最大程度地降低其使用量,那么该方案将具有较好的工业应用前景。
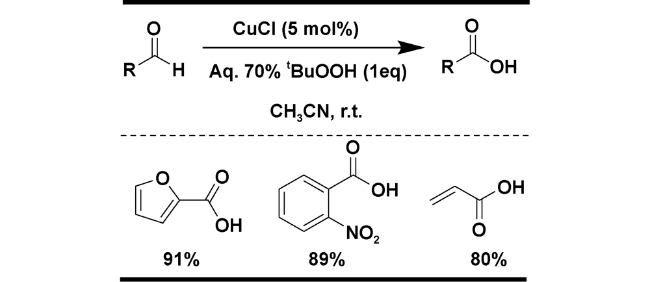
受CuII-salen配合物/30%H2O2催化体系的启发,二价铜Cu(Ⅱ)催化氧化体系引起了研究者的兴趣。Chakraborty等[19]在室温下,以70%的t-BuOOH水溶液为溶剂,在5% CuBr2的催化下,将各种芳香族、脂肪族、共轭醛类和醇类转化为相应的羧酸和酮类,其中在催化体系下反应1.5 h可以得到92%的糠酸收率(图9 ;表1 ,序号6)。
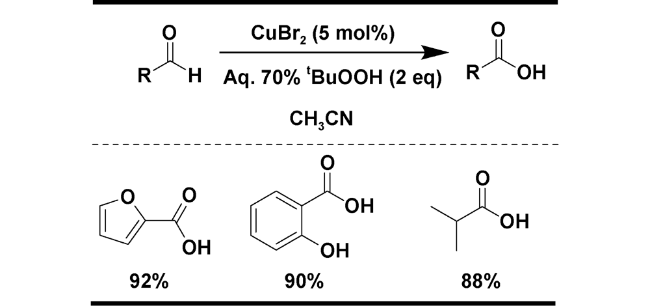
Chakraborty等[20]在醛类化合物的氧化方面做了大量的研究工作,使用催化量的Bi2O3,在70% t-BuOOH水溶液中实现了糠醛到糠酸的氧化,该方法不使用配体和其他添加剂,产物的选择性较高,不涉及烦琐的后处理和纯化操作(表1 ,序号7)。
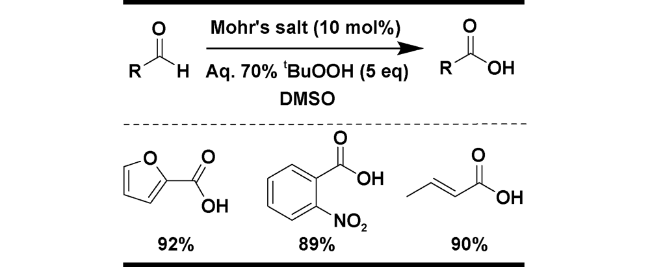
在上述工作的基础上,Chakraborty等[21]继续研究了具有催化活性以及对环境无害的过铁基催化剂(表1 ,序号8)。他们将一种常用的氧化还原指示剂[(NH4)2(Fe)(SO4)2·6H2O]氧化成Mohr’s salt用作氧化催化剂(图10 )。在催化量(10 mol%)的Mohr’s salt存在下,在DMSO、水溶剂中对H2O2、70% t-BuOOH等氧化剂进行了筛选,发现DMSO作为溶剂时得到了最优的结果。以糠醛为底物,在DMSO溶剂中,使用5当量的70% t-BuOOH水溶液作为氧化剂,80 ℃下反应3.5 h,糠醛的转化率为100%,糠酸的收率为92%,该反应体系同时适用于其他醛类底物,且具有较好的官能团耐受性。
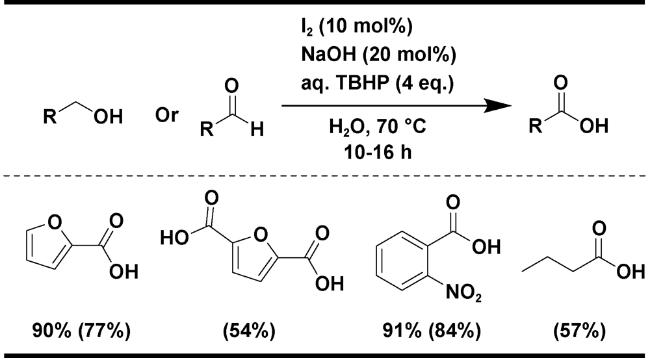
高价碘试剂即Dess Martin高碘烷(DMP)、2-碘氧基苯甲酸(IBX)和碘代苯等作为高选择性的醇到醛或是酮的当量氧化试剂,在有机合成中应用广泛。尽管DMP和IBX实用性较强且广受欢迎,但是其价格相对昂贵,且具有爆炸性,这些无法回避的缺陷限制了它们的大量使用。Elias等[22]开发了一种稳定的不含金属的I2/NaOH催化体系,使用水作为溶剂,用于催化醇或醛到羧酸的氧化(表1 ,序号9)。通过对照实验,他们发现由过氧叔丁醇(TBHP)氧化氧化碘(IO)生成的二氧化碘(IO2)物种可能是反应的活性催化物种。该方法不需要色谱纯化,适用于大规模合成,且底物兼容性比较好。使用该催化体系,以糠醛为原料得到90%的糠酸收率,以糠醇为原料可以得到77%的糠酸收率(图11 )。
2.1.4 其他氧化剂
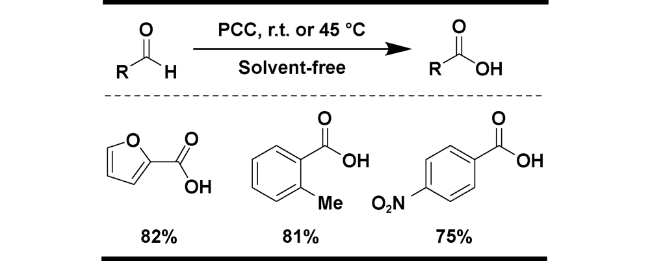
除了上述常见的氧化剂外,研究者们也对其他的氧化剂进行了研究。在无溶剂条件下,Gholizadeh等[23]报道了使用氯铬酸吡啶(PCC)可以有效地将糠醛氧化为糠酸。PCC作为一种温和的氧化剂,氧化效率高,但是其缺点是反应时间过长,且PCC本身具有毒性(图12 ;表1 ,序号10)。
Menon等[26]开发了一种二氧化碳介导的NHC催化醛到酸的方法,反应无需额外氧气的引入。二氧化碳引入过程中并未使得芳香醛转化到芳香酮酸类产物,而是生成芳香羧酸产物,机理研究表明二氧化碳在反应过程中参与了反应中间体的变化。催化体系对糠醛到糠酸的转化有明显促进作用,可以得到65%的糠酸收率(图14 ;表1 ,序号13)。
Goldberg等[27]开发具有更多给电子(六甲基苯)RuⅡ络合物催化剂,使用H2O作为氧化剂,通过醛水变换反应(AWS),在95 ℃和0.4 mol% Ru催化剂存在下,实现醛到羧酸的高效氧化(图15 ;表1 ,序号14)。研究发现使用富电子芳烃配体提高了AWS反应的选择性,而强结合的二齿辅助配体会大大降低反应性,同时,催化剂活性随着反应体系中的酸浓度的增加而降低。
从表1 汇总的结果可以看到,研究人员开发了多种均相体系,在不同的氧化剂存在下展现出不同的氧化活性。就目前已报道的体系来看,当量氧化剂的使用虽然可以取得较高的产物收率,但是原子经济性较差,三废排放较多;使用过氧化叔丁醇作为氧化剂的方法和相应的催化体系研究的比较多,但是考虑到原子经济性及工业化应用前景,开发空气或是氧气条件下的高效均相体系具有更大的吸引力,同时廉价、高效、易制备的催化剂是研究的重点;醛水交换反应在原子经济性、安全性及成本方面具有较大的优势,但是昂贵的催化剂使用、催化剂的循环套用及较低的产物收率仍是该方法面临的主要问题。
2.2 非均相氧化体系
非均相催化剂是大多数工业化生产的首选,非均相催化剂可以很容易地从产品中分离出来并且能够重复使用。表2 中汇总了近年来用于糠醛到糠酸氧化所用的非均相催化剂,具体如下。
表2 非均相催化体系催化转化糠醛到糠酸反应汇总Table 2 Summary of catalytic conversion of furfural to furoic acid by heterogeneous catalytic system |
| Entry | Catalyst | Reaction conditions | Furoic Acid Yield/% | ref |
|---|---|---|---|---|
| 1 | Au/ZTC | methanol, 6 bar O2, 120 ℃, 6 h | 90 | 28 |
| 2 | AuNPs/TiO2 | H2O, Na2CO3, 30 ℃, 8 h, UV/Uis | 92/96 | 29 |
| 3 | SiO2@Au@TiO2 | FF : Au ratio=100, 2 h, 24 bar air | 99 | 30 |
| 4 | Co4HP2Mo15V3O62 | [TEBSA][BF4], H2O2, r.t., 4 h | 94 | 31 |
| 5 | Co-N-C | H2O, 120 ℃, 1 MPa O2,1h | 96.7 | 32 |
| 6 | SiO2-Co(acac)2 SiO2-Co(acac)2 | 50 ℃, solvent-free, air, 5 h 70 ℃, H2O, air, 7.5 h | 85 180 | 33 |
| 7 | (NH4)4[CuMo6O18(OH)6] | H2O, 50 ℃, O2 balloon, 0.1 eq Na2CO3, 8 h | 99 | 34 |
| 8 | Ag2O/CuO | H2O, 70 ℃, 2 MPa O2, 1 h | 99 | 35 |
| 9 | CuO | NaOH, H2O, 65 ℃, air, 25 min | 91 | 36 |
| 10 | FeIIIMo6 | H2O, 50 ℃, 1 atm O2, 0.1 eq Na2CO3, 8 h | 97 | 37 |
| 11 | 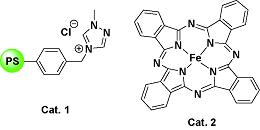 | DBU (50 mol%), anhydrous Me-THF, air, r.t. | 90 | 38 |
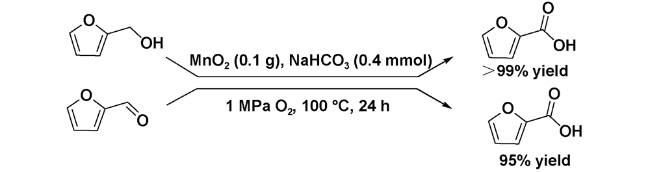
| 12 | MnO2 | H2O, NaHCO3, 1 MPa O2, 100 ℃, 24 h | 95 | 39 |
| 13 | Pd/C | Methanol-H2O, NaBH4, KOH, air, r.t. | 84 | 40 |
| 14 | [Ce(NH4)2(NO3)6] (CAN) | 1 eq. t-BuOOH (water), MeCN, r.t., 15 min | 93 | 41 |
| 15 | β- Cyclodextrin | 50 ℃, H2O2, 1 h | 97(conversion) | 42 |
| 16 | VO(acac)2-TiO2 (TSV) | H2O2, MeCN, r.t., 4 h | 86 | 43 |
| 17 | MOF-TEMPO | 1) 5 mol% catalyst, 20 mol% TBN, CH3CN, O2, 25 ℃, 2 h;2) the filtration of catalyst and additional stirring under O2 balloon at 80 ℃ for 10 h | 80 | 44 |
2.2.1 Au基催化剂
Giorgianni等[28] 通过将Au纳米颗粒沉积在由BETA沸石制备的β-ZTC碳上,获得了用于糠醛选择性氧化为糠酸的高性能Au/ZTC 催化剂,载体上仅存的弱酸位点阻碍了缩醛化和酯化途径,有利于呋喃环上醛官能团的选择性氧化到羧酸,可以实现90%的糠醛转化率和定量的糠酸选择性(表2 ,序号1)。Han等[29]发现在紫外(UV)和可见光下,Na2CO3水溶液中,使用AuNPs/TiO2催化剂可以实现糠醛到糠酸的转化。在350~400 nm 紫外灯或是420~780 nm可见光下反应 8 h,可以分别得到92%和96%的糠酸收率。同时使用糠醇作为原料,延长反应时间至48 h,可以分别在两种光源下得到90%的糠酸收率(表2 ,序号2)。催化剂循环使用性好,展现出对糠酸较高的氧化选择性。Au/TiO2催化剂制备简单,但是价格相对昂贵,开发更加廉价的非贵金属或是有机材料的光催化剂,设计更具有普适性的光催化反应器具有一定的挑战。Wojcieszak等[30]制备了核壳结构的SiO2@Au@TiO2催化剂,实现了无碱条件下的糠醛到糠酸的空气氧化。与传统负载型SiO2@TiO2@Au 催化剂相比,这些材料的催化活性提高了一百倍。研究发现核壳结构对催化剂催化活性起到关键作用,它可以防止糠酸在活性金属Au上的不可逆转吸附及催化剂失活,同时提高金属-氧化物相互作用,并且在Au-TiO2界面周边形成新的催化活性位点。在最优的反应条件下,使用0.25% SiO2@Au@TiO2催化剂,糠醛转化率达到100%,糠酸选择性达到99%以上(表2 ,序号3)。
2.2.2 Co基催化剂
Li等[31]开发了在离子液体[TEBSA][BF4]中使用的Co4HP2Mo15V3O62/H2O2催化体系用于醛类化合物到羧酸和酯的转化(表2 ,序号4)。糠酸可通过简单的有机溶剂萃取分离,催化体系可循环使用,同时该催化体系兼容多种芳香族和脂肪族醛底物,对羧酸产物有较高的选择性(图16 )。
Xu等[32]开发Co-N-C催化剂,实现糠醇到糠酸的高选择性转化,在120 ℃,氧压1 MPa,水中反应1 h可以得到96.7%的糠酸收率(表2 ,序号5)。近年来,负载的金属络合物催化剂已广泛用于有机合成中。在过去的十年中,乙酰丙酮金属配合物已成为多种反应的通用催化剂。Clark等[33]将金属乙酰丙酮化物[Co(acac)2、Cu(acac)2、Pd(acac)2、Ru(acac)3、Mn(acac)3]与有机改性的3-氨丙基二氧化硅相结合制备了一系列非均相催化剂,以研究在水溶剂中的氧化活性。他们发现,SiO2-Co(acac)2的氧化活性最高。在无溶剂、空气条件下,使用SiO2-Co(acac)2催化剂,在50 ℃反应5 h可以得到85%糠酸收率,在水溶液中,70 ℃反应7.5 h可以得到80%糠酸收率(表2 ,序号6)。
2.2.3 Cu基催化剂
Wei等[34]报道了在常压氧气条件下,以无机配体负载的Cu催化剂(NH4)4[CuMo6O18(OH)6]在水中催化氧化醛类的方法。催化剂由硫酸铜与钼酸铵在100 ℃水中通过简单的一步法合成,避免使用昂贵、有毒、空气/湿气敏感且商业上不可获得的有机配体。催化剂具有一个Cu(Ⅱ)核心,并且平面布置了六个Mo(Ⅵ)中心。在水溶剂中,氧气条件下,糠醛完全转化,以99%的收率得到糠酸。该铜基催化剂在连续使用六次后活性没有明显的降低(表2 ,序号7)。CuO 和 Ag2O/CuO催化剂能够有效地氧化糠醛制备糠酸,在最佳的反应条件下,尺寸为150 nm的Ag2O/CuO和CuO催化剂可以以接近100%的收率得到糠酸,Ag2O/CuO催化剂制备简单,成本较低,但是催化剂水热稳定性及重复性较差(表2 ,序号8)[35]。徐贤伦等[36]在水中采用不同方法合成了形貌不同的纳米CuO催化剂,其中制备的棒状CuO催化剂表现出良好的氧化活性,在糠醛一次加入的情况下,糠酸收率达91%(表2 ,序号9)。
2.2.4 Fe基催化剂
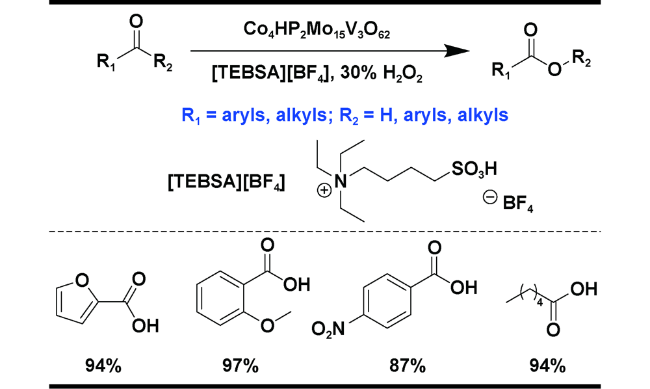
Wei等[37]制备了一种铁基杂多酸金属氧酸盐[[N(C4H9)4]3-[FeMo6O18(OH)3{(OCH2)3CNH2}](FeIIIMo6)用于水溶剂中醛的氧化反应。该催化剂是由一个单一的Fe金属支持的多钼酸盐,这种多钼酸盐是由六边共享的八面体组成的。在对催化剂活性的研究中,他们发现酸性添加剂NH4Cl的存在对氧化反应有较强的抑制作用,改变催化剂负载量对产品收率的影响不大。在50 ℃常压氧气下反应8 h,可以得到最高97%的糠酸收率。实验结果显示,该催化剂在循环使用后活性下降不明显,表现出较高的催化剂循环性能(表2 ,序号10)。Massi等[38]发展了非均相三唑钅翁催化剂和铁(Ⅱ)酞菁组成的氧化体系在醛类氧化反应中的方法(图17 )。该催化体系能够将5-羟甲基糠醛(HMF)选择性转化为5-羟甲基-2-呋喃甲酸(HMFCA),将糠醛高效氧化到糠酸,收率为90%(表2 ,序号11)。还可以将HMF和糠醛直接转化为其相应的酯、酰胺和硫酯等衍生物,这进一步说明非均相N-杂环卡宾催化剂在生物质领域中的应用潜力。
2.2.5 其他催化剂
强碱的存在有利于羧酸产物的获得。Chakraborty等[41]使用1 mol%的硝酸铈铵[Ce(NH4)2(NO3)6] (CAN)作为催化剂,在乙腈溶剂中室温条件下,使用70% t-BuOOH水溶液作为氧化剂,反应15 min可以得到93%糠酸收率(表2 ,序号14)。
沈静茹等[42]使用新型β-环糊精衍生物作为催化剂,在50 ℃,酸性条件下,反应1 h可以将糠醛氧化到糠酸,转化率为97%,催化剂可以在丙酮等有机溶剂中析出进而回收套用,催化剂重复使用4次,催化剂的活性没有明显的下降(表2 ,序号15)。Thakur 等[43]将VO(acac)2负载在TiO2上制备非均相TSV催化剂,在双氧水存在下,催化剂表现出对糠醛氧化制备糠酸或是糠酸酯较好的催化活性,在室温下反应4 h,乙腈中可以得到86%的糠酸收率,甲醇中可以得到75%的糠酸甲酯收率(表2 ,序号16)。Kim等[44]开发一种TEMPO功能化的金属有机骨架 (MOF) 催化剂,通过非均相MOF-TEMPO催化剂的添加和移除,实现醇一锅法到羧酸的氧化(图19 )。通过该方式的引入,可以有效地解决TEMPO催化醇的好氧氧化及醛在自由基抑制下难以自氧化难题,有效串联两步氧化过程,实现多种羧酸产品的制备(表2 ,序号17)。
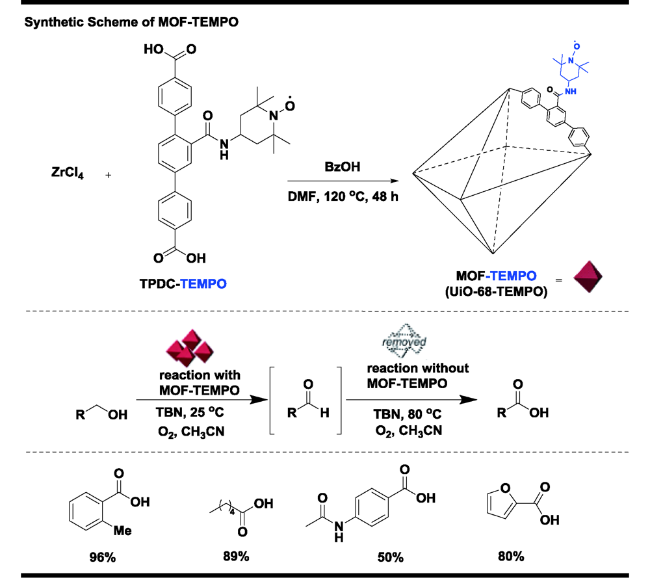
从表2 中可以看出,总体而言,在非均相催化剂的研究中,报道了许多负载型贵金属催化剂,如SiO2@Au@TiO2、AuNPs/TiO2、Pd/C和Ag2O/CuO催化剂等,这些催化剂的生产成本较高。纳米氧化铜易于制备,成本较低,但是催化剂重复使用性较差,反应需要在一定量的碱(如氢氧化钠)存在下方可以保持催化剂的稳定,而碱的使用会造成后处理过程中产生大量盐,对环境造成污染,所以避免碱的使用是十分重要的。在最近的研究中,Co4HP2Mo15V3O62体系使用离子液体作为溶剂,但是溶剂的成本比较高。此外,t-BuOOH作为氧化剂,产物收率较高,反应时间较短,不需要额外碱添加,但是t-BuOOH的分离纯化比较难,大规模使用成本较高,且存在一定的安全隐患。具有核壳结构的负载催化剂在催化剂稳定性及活性上有较大的优势,开发廉价的核壳活性金属催化剂是未来研究的重要方向。通过绿色氧化技术来实现糠醛到糠酸的转化仍然具有一定挑战,开发新型非均相催化体系来实现重要的生物基平台分子有氧氧化制备高附加值化学品仍然具有重要的研究价值。
2.3 生物氧化法
生物转化通常在温和的反应条件下进行,生物催化剂具有极好的选择性、可生物降解性和环境友好性,生物催化氧化法作为一种有前途的选择,可以补充甚至替代化学方法。
Li等[45]报道了使用新分离的菌株 Comamonas testosteroni SC1588 将呋喃基平台分子生物催化氧化为相应羧酸的方法。与生长中的细胞相比,休眠细胞被发现是更好的生物氧化催化剂。该菌株表现出对糠醛类化合物的高耐受性(高达 180 mmol/L)。在50 mmol/L 糠醛、30 mg/mL微生物细胞、4 mL 磷酸盐缓冲液(200 mmol/L,pH 7.0)、20 mmol/L组氨酸、30 ℃下反应96 h可以得到最高93%的糠酸收率。由于该菌株高的解毒效率和呋喃醛基耐受性,可能在木质纤维素水解物的生物降解中具有广阔的应用潜力,同时它不能同时利用葡萄糖和木糖作为碳源,从而避免了生物解毒过程中的糖损失。PÉrez等[46]利用Nocardia corallina B-276全细胞,采用分批培养与静止细胞在磷酸钾缓冲液(0.1 mol/L,pH 7.0)中研究糠醇和糠醛氧化制备糠酸的反应。在3 L生物反应器中,2-糠醇(1.35 g/L)在24 h后被转化为糠酸,收率为81%,利用静止细胞进行2-糠醇的转化,在21 h内2-糠酸的产量达到98%。在底物与细胞的比例为1∶3.5(w/w)时,反应8 h,糠醛被生物氧化可以得到88%的糠酸收率。de María等[47]使用市售固定化脂肪酶Candida antarctica (Immo-CAL-B)作为生物催化剂,在50 mmol/L糠醛, 10 mg/mL CAL-B催化剂,乙酸乙酯:叔丁醇= 1∶1 (v/v)的混合溶剂中,逐小时加入共9.6当量的过氧化氢水溶液(30 % v/v),原位形成过酸,随后氧化糠醛得到91%的糠酸收率。其中烷基酯作为酰基供体。
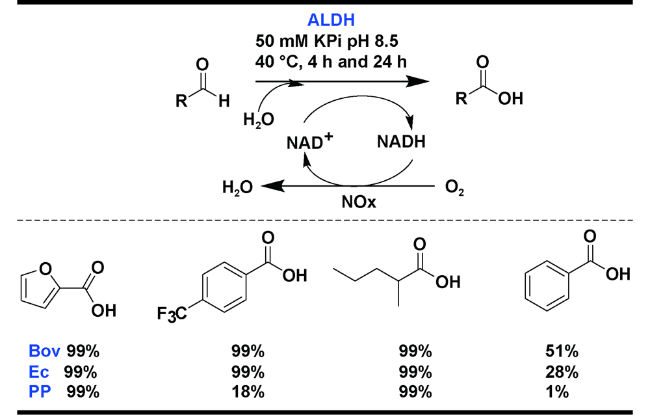
Mutti等[48]使用源自细菌和牛晶状体的三种重组醛脱氢酶(ALDH-Bov、ALDH-Ec和ALDH-PP)对醛进行了氧化(图20 )。这些酶以高度纯化的形式与烟酰胺氧化酶(NOX)结合使用,消耗空气中的分子氧得到催化性的 NAD+物种。他们使用冻干的全细胞和静止细胞生物催化剂研究了该反应。研究表明羧酸产物是唯一被检测和分离得到的,化学选择性超过99%。该方法底物兼容性好,避免使用有毒或不安全的氧化剂、强酸及强碱同时也无其他经典的副反应(例如不饱和官能团的卤化、Dakin 型氧化等)。醛脱氢酶是醛化学选择性氧化成羧基的重要催化剂,未来的研究将集中在提高酶对底物浓度的耐受性和长期稳定性,以使这些酶得到更广泛的应用。
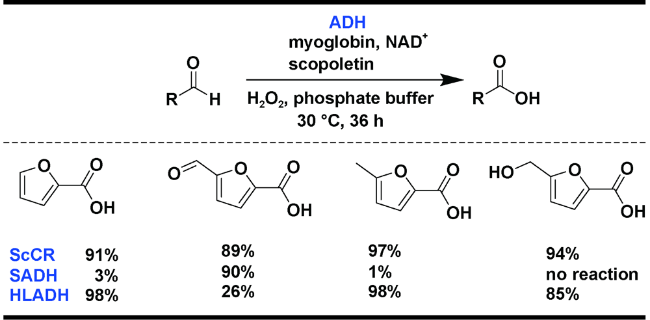
Li等[49]报道通过重组大肠杆菌细胞从相应的醛中选择性合成呋喃羧酸,该大肠杆菌细胞表达来自睾酮假单孢菌SC1588 的3-琥珀酰半醛-吡啶脱氢酶 (SAPDH)。研究了诱导和反应条件对糠醛(FF)全细胞催化氧化的影响。他们发现通过使用高温诱导,糠酸的产率和选择性显著提高,但反应时间稍长,该重组菌株在较窄的 pH 范围(7~8)内表现出良好的催化活性。该重组菌株对糠醛的耐受水平约为 100 mmol/L,糠酸的产率为 97%。Li等[50]构建了由半乳糖氧化酶 (GOase) 和醇脱氢酶 (ADHs) 组成的双酶级联系统,筛选的ADHs 包括来自Streptomyces coelicolor 的羰基还原酶 (ScCR)及来自Synechocystis sp.和horse liver 的 ADH酶 (SADH和 HLADH)。这种双酶级联氧化系统具有较低的O2依赖性、更高的原子效率和产物可控性,对多种呋喃基底物有优异的普适性且无需添加过氧化氢酶(图21 )。Li等[51]通过辅因子工程化的大肠杆菌(Escherichia coli)细胞生物氧化醛底物合成羧酸产物。他们将NADH氧化酶(NOX)引入到含有醛脱氢酶(ALDH)的大肠杆菌中,以促进细胞内NAD+的再生,从而显著增强ALDH催化氧化能力。这些工程化的生物催化剂能够有效地氧化各种芳香醛。更重要的是,它们对有毒性的呋喃底物表现出很高的底物耐受性。大肠杆菌、香草醛脱氢酶和NOX共表达 (E. coli_CtVDH1_NOX),能够将大于240 mmol/L的糠醛和5-羟甲基糠醛(HMF)高效氧化为2-糠酸和5-羟甲基-2-糠酸,生产效率达到2.0和3.7 g/Lh。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
生物催化氧化法虽然取得了许多显著和可喜的成果,但要实现具有经济性的糠酸规模化生产还需进一步改进。由于呋喃醛的毒性可强烈抑制生物催化剂的活性,因此开发更高耐受性和稳定性的生物催化剂并将其应用于糠酸及相关产物的生产过程中具有重要意义。其次,为实现糠酸的高效生产,应进一步优化级联酶反应过程,连接上下游工艺,避免中间产物的积累,简化下游提取过程。同时应通过酶工程和基因工程的随机或定点诱变和表达系统的优化,系统地设计和构建具有高活性和高产量的高效突变体。为了降低生产成本,需要对酶和全细胞固定化方法进行研究,以获得经济上可行的、稳定、活性高、可重复使用的固定化生物催化剂。
3 结论与展望
目前,糠醛氧化制备糠酸应着力于开发温和、廉价且可以在无碱环境下使用的新型非均相催化体系,我们认为,加大以下方面的研究可能会对糠酸的绿色合成有一定的启发:
(1)发展环境友好催化体系。开发高效、低成本、稳定的催化剂来实现有氧氧化是非常必要的。与贵金属催化剂相比,非贵金属催化剂用于有氧氧化的研究较少,尽管取得了一定结果,但是还需要进一步研究。此外,应当主要发展以空气为氧化剂的催化体系。与其他氧化剂相比,空气有许多优点,如高原子经济性且副产品只有水。然而,以分子氧作为终端氧化剂的廉价高效催化方法仍然缺乏。因此,在水相体系中利用分子氧作为氧化剂的非贵金属催化剂是值得探索的。
(2)催化剂的稳定性至关重要。在已经报道的有机金属催化剂的例子中,大量使用了各种金属配体、膦配体或氮杂环卡宾,这些配体都易受氧化自我降解,这是过渡金属催化过程中经常遇到的问题。此外,催化剂的稳定性取决于活性金属种类与载体之间的相互作用。在反应条件下,载体应该是稳定的。从现有的研究中可以清楚地看出,载体的作用在实现高转化率和高选择性方面起着决定性的作用。通过催化剂结构的设计和开发,提高非均相催化剂的水热稳定性,尤其是在无碱条件下的稳定性是非均相催化剂亟待解决的问题。
(3)采用光催化、电催化、超声处理、微波加热以及生物法等催化方法实现糠醛的无碱氧化是值得进一步探索的替代方案。绿色化学的基本原理是促进可再生原料与更安全的溶剂和助剂的使用,以发展危险性更低的化学合成。在较温和的条件下,利用多种新型的催化方法来提高工艺效率,防止废弃物的产生势在必行。