



1 引言
质谱通过准确测定离子的质量-电荷比(mass-to-charge, m/z)来推导分子结构。这是一种普适性谱学方法,由于其高灵敏度、高分辨率和高特异性等特点而广泛应用于鉴定小分子药物、蛋白质、核酸、脂质及糖类物质结构[1⇓⇓~4]。由于质谱是对带电荷粒子的检测,发展新型电离(Ionization)/解离(Dissociation)技术是研发质谱仪器和分析方法的关键。鉴于解离和电离的紧密联系,本文将在综述各种电离技术的基础上,分析由各种电离技术所产生的分子离子性质、特点和进一步解离途径,然后重点阐述光解离(Photo Dissociation)及其与各种软电离(Soft Ionization)技术的联用。
2 质谱电离方法概述
目前质谱广泛采用的电离技术大都基于电子得失、质子化/去质子化或形成金属离子加合物等原理,表1 概括了电子轰击/俘获电离、电喷雾电离、基质辅助电离、表面电离和原子/离子束电离等主要电离技术的特点和应用。
2.1 电子轰击/俘获电离
1918年Dempster建立的电子轰击电离(Electron Impact Ionization, EI)是最经典的电离方法,广泛与气相色谱联用。在这种方法中,灯丝发射的热电子被加速,高能电子束与中性分子碰撞,致使其丢失低电离电位电子,产生具有高反应活性的阳离子自由基,并进一步发生由自由基中心引发的化学键均裂或正电荷中心引发的化学键异裂,或伴随分子重排。高能电子轰击方法(一般采用70 eV电子束)[5]通常被称为“硬电离”,因为70 eV电子的de Broglie波长大约为0.14 nm,与一般化学键键长相当,这种高能电子与分子之间的强相互作用,常常引起振动激活的非特异性化学键断裂,影响分析谱图中的碎片离子。2012年钟鸿英等报道的低能电子俘获是一种新型软电离方法,即激光诱导隧道电子俘获(Laser Activated Electron Tunneling, LAET)[6⇓⇓⇓~10],这种方法利用半导体表面在激光照射下产生的光电子来实现中性分子的电离和解离。在静电场中,光激励的电子从半导体价带跃迁到导带,界面转移光电子被吸附在半导体表面样品分子中的缺电子原子俘获,产生具有一个未配对电子的阴离子自由基,并进一步发生由自由基中心或负电荷中心引发的解离。与高能电子轰击相反,电子俘获是一个可以自发进行的放热过程,因此不需要高能量,低能电子(<20 eV)俘获有效减少了振动激活的非特异性化学键断裂。这两种由电子得失引发的电离方式均产生具有未配对电子的自由基阳离子或自由基阴离子,这种类型的分子离子均不稳定,无需其他额外装置便可进一步发生自由基或电荷中心引发的化学键断裂,从而获得结构鉴定所需的特征碎片离子。
2.2 电喷雾电离和基质辅助电离
1984年Fenn等报道的电喷雾电离(Electrospray Ionization, ESI)[11]和1988年Tanaka等报道的基质辅助激光解吸离解(Matrix Assisted Laser Desorption Ionization, MALDI)[12]可通过质子化/去质子化,或与阳离子(如金属离子Na+、K+和Li+等)形成加合物等途径来实现中性分子的电离。相对于硬电离技术,这种软电离方法对样品破坏小,广泛应用于生物大分子分析,特别是蛋白质氨基酸序列和翻译后修饰分析[13⇓⇓~16]。这两种软电离技术大大推进了生物医学研究进程,Tanaka获得了2002年诺贝尔化学奖。与电喷雾电离/基质辅助电离相似,1995年由Lindinger等建立了质子转移/电荷还原(proton transfer/charge reduction, PTR)[17]技术,主要用于分析质子亲合性高于水的挥发性气态有机小分子。该技术利用空心阴极对水蒸气放电,生成的高纯度H3O+在漂移管发生分子-离子反应,可用于实时在线检测大气痕量有机污染物质,也可用于辅助诊断肺癌等疾病[18]。这三种方法所产生的准分子离子均为偶电子离子,比具有奇数电子的阳离子/阴离子自由基更稳定,一般很难自发进行解离,因此谱图中碎片离子较少。为了获得更多结构信息,需要与其他解离技术联用,将这些准分子离子进一步裂解成碎片离子。
2.3 表面电离
1993年Bill Hutchens和Tai Ying Yip提出的表面增强激光解吸电离(Surface Enhanced Laser Desorption Ionization, SELDI)[19]是在基质辅助电离基础上发展起来的表面电离方法,将MALDI与蛋白芯片(protein chip)[20]相结合,利用化学(离子交换、反相/正向分离或金属离子配位等)或生物化学(抗体或受体等)技术改性处理样品靶表面,选择性地结合样品中目标分子,并采用适当溶剂清洗除去其他共存成分。芯片上保留的蛋白则与能够吸收激光能量的有机小分子基质共结晶,在特定波长激光照射下,待测目标分子通常从质子化途径实现电离。这种蛋白芯片SELDI技术具有高度集成、微型、自动化和高通量等特点,与飞行时间质量分析器联用可广泛应用于疾病标志物筛选、早期诊断和新药研发等领域[21,22]。
由于MALDI使用的有机小分子基质在低质量端产生较大背景干扰,而且激光热效应使得实际解离面积大于激光光斑面积,影响质谱分析的空间分辨率。Buriak等于1999年报道的多孔硅表面解析离子化技术(Desorption Ionization on Porous Silicon, DIOS)[23]是一种新型无基质表面电离技术,这种技术利用具有纳米结构的多孔硅吸收激光能量并使待测分子离子化,有效克服了 MALDI基质干扰问题,可用于小分子代谢物的质谱分析[24⇓~26]。2007年Siuzdak等报道了另外一种无基质的表面电离技术,这种被称为纳米结构启动子的质谱离子化技术(Nanostructure-Initiator Mass Spectrometry, NIMS)[27]在多孔硅表面的“笼形”(clathrate)纳米结构中聚集启动分子氟化物比如双(十三氟辛基)四甲基二硅氧烷(bis(tridecafluoro-1,1, 2, 2-tetrahydrooctyl)tetramethyldisiloxane),当受到离子束或光照射时,启动分子受热挥发,使吸附在表面的样品分子离子化。由于启动分子非常稳定,不产生背景离子干扰。这种软电离技术不仅可用于小分子代谢物、药物、蛋白质和多肽等分析,也可用于细胞和组织成像。由于样品前处理简单,可直接进行血液和尿样分析[28⇓⇓~31]。这些表面电离方式均为软电离技术,产生的准分子离子为稳定性较高的偶电子离子,一般也不能自发进行化学键断裂,需要借助其他解离技术来进一步产生碎片离子。
2.4 原子/离子束电离
离子束电离技术包含两类,一种属于硬电离技术,另一种属于软电离技术。二次离子质谱(Secondary Ion Mass Spectrometry, SIMS)[34]使带有几千电子伏特能量的离子束(Ga+、A 、B 和Cs+等)轰击样品表面,属于硬电离技术,其间伴随一系列物理和化学变化,包括离子束在样品表面的小概率被散射、样品粒子溅射和吸附层表面的化学反应等。轰击样品的一次离子在穿透固体样品表面过程中与晶格原子发生弹性或非弹性碰撞,晶格原子获得能量后,部分移向表面并把能量传递给表面样品,溅射出原子、原子团、元素离子、碎片离子或分子离子等正负离子,或通过表面化学反应产生二次离子。利用SIMS可进行样品表面或内部化学元素和化合物鉴定、固体材料或生物组织和细胞表面成像等[35]。通过对样品表面进行交替溅射剥离和溅射区域图谱采集,还可以对不同成分进行深度剖析,获得化学成分微区分布的三维图像[36⇓~38]。二次离子质谱具有很高的灵敏度,可低至ppm甚至ppb,空间分辨率低至1 nm[39]。但是离子束的能量高达几千电子伏特,引起振动激活的非特异性化学键断裂,产生大量碎片离子,难以获得完整生物分子的准分子离子。按照离子束流密度大小,SIMS分为静态SIMS(Static-SIMS,>1013离子/cm2)和动态SIMS(Dynamic-SIMS,>10离子/cm2),分别与飞行时间(Time-of-Flight, TOF)和双聚焦质量分析器联用。自1979年德国科学家Benninghoven研制出第一台TOF-SIMS[40]以来,现在已经发展到第五代TOF-SIMS。在此期间,发展出了不同离子源以适应不同分析目的,包括气体放电源、表面电离源、热隙源和液态金属及团簇源等[41],广泛应用于材料、能源和生物医学等领域。利用电荷补偿原理的电子中和枪,SIMS甚至还可以分析绝缘样品[42]。
2005年由Laramee和Cody发明的实时直接分析质谱(Direct Analysis Real Time, DART)[47]是一种表面非接触型常压气态原子/离子束混合质谱电离技术。在大气压条件下,中性或惰性载气(如氮气或氦气)经放电产生离子、电子和长寿命电子激发态原子或分子,通过电离区时大部分离子和电子被除掉,剩下长寿命的电子激发态原子或分子。当载气为氮气时,这些激发态原子或分子束冲击样品时与空气中的水分子发生潘宁电离,产生水合物离子簇和电子。在正离子模式下,水合物离子簇与样品待测物分子发生瞬间质子交换反应,产生质子化阳离子。在负离子模式下,潘宁电离产生的热电子与大气中的氧气和水分子反应,产生的氧气和水分子离子化簇与待测物分子反应,并产生去质子化负离子。DART与DESI均为常压软电离技术,溶剂和高盐样品对DART影响小,特别是不产生多电荷离子和金属离子(比如Na+、K+和Li+)加合物,谱图简单容易识别,可用于定性结构鉴定和定量分析[48⇓⇓~51]。
综上所述,基于不同物理化学原理的电离方法各有特点,适用于不同分析目的。除了激光诱导隧道电子俘获软电离(LAET)、电子轰击硬电离(EI)以及二次离子硬电离(SIMS)技术,其他几种常见的软电离技术(如ESI和MALDI)均产生较稳定性的偶电子离子,需要与其他解离技术联用才能实现分子离子的进一步解离。
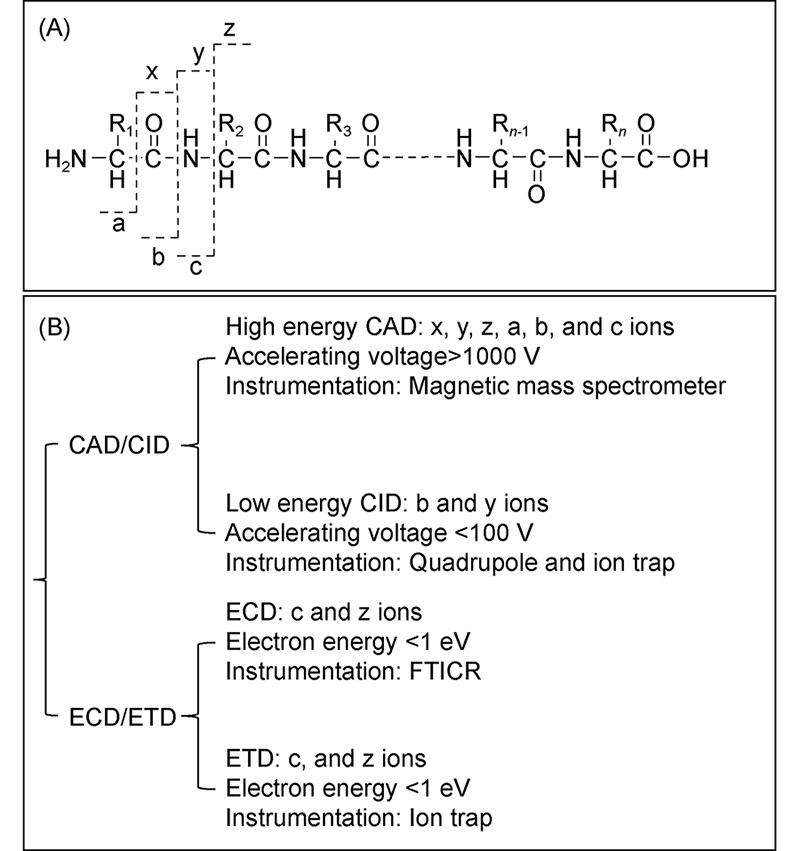
3 质谱解离技术概述
碰撞活化、电子得失和光辐射是质谱中常见的三种解离方式,这些方法利用不同原理产生不同碎片离子。
碰撞活化解离(Collision Activated Dissociation, CAD)或碰撞诱导离解(Collision Induced Dissociation, CID)[52⇓⇓⇓⇓⇓~58]是串联质谱(Tandem MS/MS)中最经典的离子解离技术。带有一定能量的离子在碰撞室中与情性气体分子或原子发生碰撞,引发碰撞激活的化学键断裂,产生碎片离子。离子的动能大小直接影响碰撞活化解离效率,磁式质谱仪一般采用高能CAD[59],离子加速电压超过1000 V。四极杆和离子阱等一般采用低能CID,加速电压不超过100 V[60]。两者得到不同碎片离子谱,比如,在正离子模式下,高能CAD使多肽骨架中C—C、C—N和N—Cα三种不同化学键断裂,所得谱图包含所有x、y、z、a、b和c碎片离子,而低能CID一般主要形成b和y 离子(图1 )。CAD或CID为接近热力学平衡条件下的各态振动能级历经(ergodic)过程,即吸收的能量按照热力学统计规律分布于所有振动能级,化学键断裂缺乏选择性,与蛋白质共价结合的糖基、磷酸基团和脂基等的解离与主链化学键断裂同时发生,导致丢失与翻译后修饰相关的重要碎片离子信息。不仅如此,对非胰蛋白酶剪切肽段、高质量长肽段和高碱性肽段等阳离子,碰撞活化/诱导难以使化学键有效断裂并产生碎片离子。
在阳离子中引入电子,使稳定的偶电子离子转变为不稳定的奇电子离子是提高解离效率的重要途径。1998年由Zubarev和McLafferty等提出的电子俘获解离(Electron Capture Dissociation, ECD)[61]以及2004年由Hunt和Syka建立的电子转移离解(Electron Transfer Dissociation, ETD)均为远离热力学平衡的非各态历经(non-ergodic)解离技术。在ECD技术中,低能自由电子(通常小于1 eV)与质子化蛋白质或肽离子相互作用,当电子与正离子接近时,静电库仑力使电子被蛋白质或肽阳离子捕获,将其由偶电子离子转变为奇电子离子,并进一步发生自由基引发的 N—Cα键断裂,形成 c 和z 类型离子(图1 )。这种方法增强了非胰蛋白酶剪切肽段、长肽段和高碱性肽段产生碎片离子的能力,并保留了较弱的翻译后修饰基团。但是阳离子与自由电子的共存面临较大困难,使ECD在蛋白质组学中的广泛应用受到仪器限制,只能用于FTICR-MS[62⇓⇓~65]。针对这个问题,ETD技术改由化学电离方法(Chemical Ionization, CI)产生携带电子的阴离子(anion)[66⇓~68],阴离子与蛋白质或肽阳离子相互作用,引发与ECD相似的化学键断裂。ETD技术已广泛应用于线性离子阱质谱仪,可在100~300 ms内产生碎裂离子,适合高通量蛋白质组学分析[69]。与ECD技术一样,ETD技术能够较好地选择性断裂主链,不但可以获得丰富的c、z序列离子,而且能够较好地保留翻译后修饰基团包括磷酸化修饰[70]、N-和O-糖基化修饰[71]、磺化修饰[72]等,为蛋白质氨基酸序列和翻译后修饰分析提供了有效手段。ETD技术显著提高了对携带多电荷的大肽段或完整蛋白[73]、高碱性蛋白比如组蛋白(histone)等的分析能力,在top-down[74]蛋白质组分析和de novo[75]测序等方面具有较大的应用前景。
与上述电离/解离方式相比,光电离/解离技术利用波长/能量可调控的光辐射来使样品分子电离,并激活特定化学键断裂,本文将重点从基本原理、仪器特点及其在生物分子(包括有机小分子,蛋白质、核酸和糖)结构鉴定中的应用等方面综述近年研究进展。
4 光电离/解离基本原理
在不同波长光子(X射线、紫外光、红外光和微波)辐照下,样品分子或原子(气体、液体或固体)可通过多种渠道发生电离。当光子能量大于电离能时,分子或原子吸收光子而直接电离是主要的光电离渠道。其中红外和微波则需经过分步激发途径,使原子和分子逐步进入高激发态而电离。光电离/解离的最终产物离子取决于多光子吸收过程与电离、能量弛豫和解离过程的竞争进行,碎片离子种类取决于激光强度、波长和脉冲宽度。当脉冲宽度和激发波长一定时,可以非常方便地通过调节激光强度来控制解离程度。除此之外,受激发的原子与分子还可以通过与电子、离子、中性粒子以及同种粒子之间碰撞而电离。
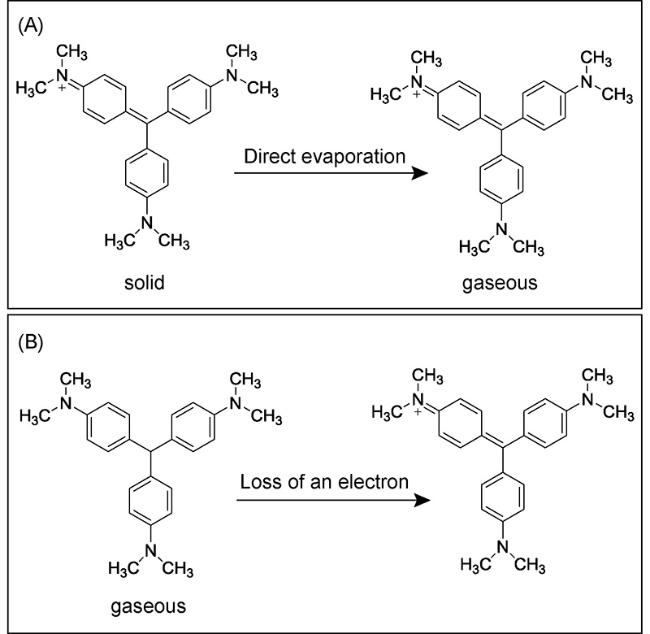
图2 激光解吸电离甲基紫的两种途径。(A)激光热效应直接气化固态甲基紫盐酸盐;(B)激光激励中性甲基紫丢失低电离电位电子Fig.2 Laser desorption dissociation of methyl violet. (A) Direct evaporation of solid methyl violet hydrochloride by laser heating effect; (B) Losses of low ionization potential electrons of neutral methyl violet by laser excitation |
4.1 直接光电离和解离
激光解吸电离是光电离现象的早期应用实例,当一定波长和脉冲宽度的激光直接照射到样品上,中性分子吸收光子后发生光电离(Photo Ionization, PI),丢失低电离电位电子,产生阳离子自由基。若吸收多个光子,获得足够的能量,还可进一步发生光电离/解离(Photo Ionization Dissociation, PID),即发生自由基中心引发的化学键均裂、电荷引发的化学键异裂或振动激活的非特异性化学键断裂,产生正离子和负离子。如果样品本身为正离子比如盐酸甲基紫,如图2 (A)所示,激光的热效应使其汽化,发生光解吸(Photo Desorption, PD)并进入质量分析器。激光解吸电离(Laser Desorption Ionization, LDI)没有基质干扰,可用于有机小分子分析[76⇓⇓⇓~80]。图2 (B)是圆珠笔主要染料甲基紫(Methyl Violet)的结构式及光电离机理,这个分子具有能够吸收激光(337 nm)的基团,成键轨道上的电子吸收光子跃迁至反键激发态轨道,激发态分子丢失电子生成m/z 372阳离子自由基离子。因为电荷可以被分散离域于大环共轭体系,该准分子离子具有较高的稳定性,在质谱图中为基峰,较少产生碎片离子。LDI技术要求样品分子具有能够吸收激光能量的基团,由于激光直接照射样品分子,因此只适用于光稳定的样品。相比之下,基质辅助激光解吸离解(MALDI)更为普遍,利用有机小分子作为基质与样品分子共结晶,既起到吸收激光能量和包裹保护样品作用,又能提供质子使样品分子质子化,将待测物中性分子转化为阳离子。
表1 质谱中不同电离技术的原理及应用Table 1 Principle and application of different ionization techniques in mass spectrometry |
| No. | Ionization | Principles and products | Molecular ions | Fragment ions | Whether the coupling to other techniques for fragmentation is needed (Y/N) | Applications | |
|---|---|---|---|---|---|---|---|
| 1 | Electron-initiated ionization | Electron impact (EI) | Loss of electrons with low ionization potential Radical cation | Ions with odd-numbered electrons | Radical/charge-initiated homolytic or heterolytic bond cleavages | N | Volatile small organic molecules |
| Laser activated electron tunneling (LAET) | Electron capture by charge deficient atoms Radical anions | Ions with odd-numbered electrons | Radical/charge-initiated homolytic or heterolytic bond cleavages | N | small organic molecules | ||
| 2 | Electrospray ionization (ESI) | Protonation/deprotonation Metal ion adducts | Ions with even-numbered electrons multiple-charged ions | Difficult to occur spontaneously | Y Collision-activated dissociation, electron transfer dissociation and photo dissociation | biological macromolecules/small organic molecules | |
| 3 | Matrix assisted laser desorption ionization (MALDI) | Protonation/deprotonation Metal ion adducts | Ions with even-numbered electrons | Difficult to occur spontaneously | Y Collision-activated dissociation and photo dissociation | biological macromolecules | |
| 4 | Surface ionization | Surface Enhanced Laser Desorption Ionization (SALDI) | Protonation/deprotonation Metal ion adducts | Ions with even-numbered electrons | Difficult to occur spontaneously | Y Collision-activated dissociation and photo dissociation | biological macromolecules/small organic molecules |
| Desorption Ionization on Porous Silicon (DIOS) | Protonation/deprotonation Metal ion adducts | Ions with even-numbered electrons | Difficult to occur spontaneously | Y Collision-activated dissociation and photo dissociation | biological macromolecules/small organic molecules | ||
| Nanostructure-Initiator Mass Spectrometry (NIMS) | Protonation/deprotonation Metal ion adducts | Ions with even-numbered electrons | Difficult to occur spontaneously | Y Collision-activated dissociation and photo dissociation | biological macromolecules/small organic molecules | ||
| 5 | Atomic/ion beam ionization | Fast Atom Bombardment (FAB) | Protonation/deprotonation Metal ion adducts | Ions with even-numbered electrons | Difficult to occur spontaneously | Y Collision-activated dissociation | Polypeptide/small organic molecules |
| Secondary Ion Mass Spectrometry (SIMS) | Positive/negative ions | Ions with even-numbered electrons | Vibration activated dissociation | N | Material elements/small organic molecules/insulators | ||
| Desorption Electrospray Ionization (DESI) | Protonation/deprotonation Metal ion adducts | Ions with even-numbered electrons multiple-charged ions | Difficult to occur spontaneously | Y Collision-activated dissociation, electron transfer dissociation and photo dissociation | biological macromolecules/small organic molecules | ||
4.2 光解离与其他电离技术的联用
对其他电离技术产生的稳定偶电子离子,如质子化阳离子和金属离子加合物,如果采用各态历经的高能碰撞活化解离(CAD)技术,往往造成振动激活的非特异性化学键断裂。近年来发展起来的光解离技术,利用特征官能团的选择性光吸收,能够提高化学键断裂的选择性。根据所使用的波长,光解离技术主要应用在红外区和紫外区[81]。
红外多光子解离(Infrared Multiphoton Dissociation, IRMPD)是光解离技术的典型,对那些CAD技术难以测定的蛋白质翻译后修饰特别是磷酸化(Phosphorylation)[82,83]、磺基化(Sulfation)修饰[84]等,IRMPD通过连续吸收多个光子,使偶电子准分子离子逐步进入高激发态,发生特定波长红外光振动激活的化学键断裂,这是一种“slow heating”碎裂原理[85]。以质子化二甘醇二甲醚阳离子为例[86],图3 比较了电子轰击电离和在944 cm-1红外光辐射下所发生的光解离。图3 (A)显示在电子轰击下,中性分子丢失电离电位低的电子成为阳离子自由基m/z 134,由于该离子不稳定,这个分子离子难以检测,由自由基中心和正电荷中心引发化学键均裂和异裂,分别得到m/z 59和89碎片离子。由于C—O—C伸缩振动在红外指纹区1150~900 cm-1有强吸收,因此在944 cm-1红外光辐射下,C正电荷中心相邻C—O键发生振动激活异裂,如图3 (B)所示,两个正电荷中心分别产生m/z 59和103正离子。不仅如此,m/z 103的正离子还可继续吸收红外光,发生相似化学键断裂,同样得到m/z 59正离子。实验还发现,在933~953 cm-1范围内激发光波长对裂解途径没有影响,但是影响解离效率。944 cm-1处激光辐射时间直接影响碎片离子强度,辐射时间越长,m/z 59碎片离子峰越强。可见,红外多光子解离技术基于特定官能团对特定波长辐射的吸收,选择性增强了阳离子正电荷引发的化学键异裂。
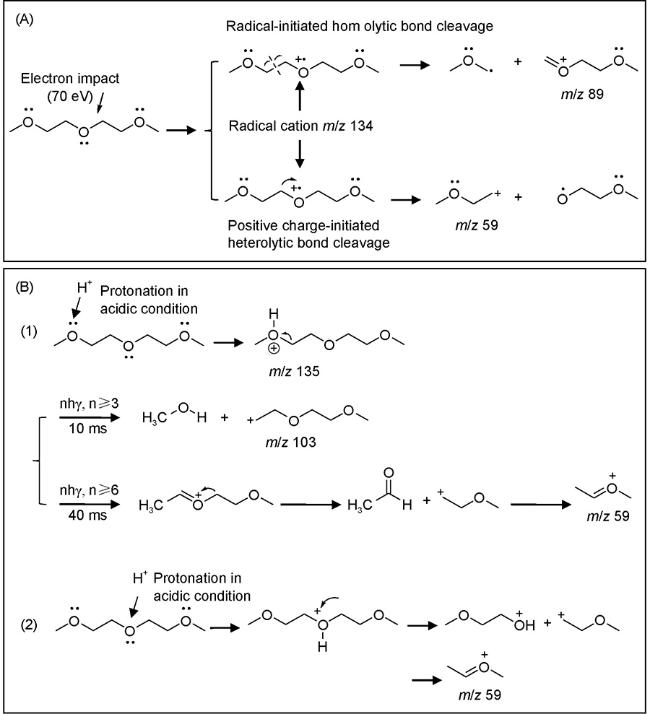
图3 红外多光子解离与电子轰击电离的比较。(A)电子轰击产生的自由基中心和正电荷中心及其引发的化学键断裂;(B)红外多光子振动激活的化学键断裂Fig.3 Comparison of infrared multiphoton dissociation and electron impact ionization. (A) Radical/charge centers generated by electron impact ionization and resultant chemical bond cleavages; (B) Infrared multiphoton activated chemical bond cleavages |
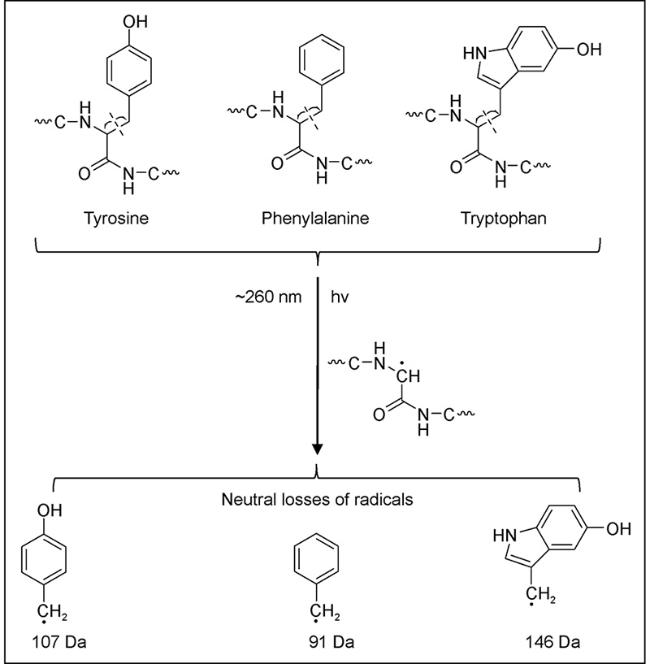
每个紫外光子的能量远远大于红外光子,分子吸收紫外光子后可在纳秒级时间内积累能量达到电离阈值,发生单光子引发的紫外光解离(Ultraviolet Photo Dissociation, UVPD)。这种解离大大降低了离子碰撞失活,解离效果类似 HCD(high energy collision induced dissociation)。紫外光解离按紫外光的波长分类也分为真空紫外光解离(Vacuum Ultraviolet Photo Dissociation, VUVPD)和近紫外光解离,近紫外光解离最常用的紫外光波长是 266 nm(4.61 eV)和 355 nm(3.49 eV),260 nm 附近紫外光可被芳香氨基酸所吸收,包括色氨酸、酪氨酸和苯丙氨酸等。如图4 所示,含酪氨酸残基多肽可发生特异性的 Cα—Cβ 侧链均裂,产生一个107 Da 的中性丢失[87]。近紫外光解离应用较少,因为许多生物分子不能直接吸收近紫外光,需要引入生色团。真空紫外光解离具有更高光子能量,能被大多数生物分子吸收,因此得到了更加广泛的应用[88⇓~90]。常使用的157 nm[91,92]和 193 nm[93] 激光可分别提供高达7.9 eV 或 6.4 eV 的光子能量,但需在真空条件下使用,以避免空气对光的吸收。
5 光解离质谱仪器的结构及特点
光解离技术常常与电喷雾电离和基质辅助激光电离技术联用,增强其产生特征碎片离子的能力。如前所述,中性样品分子经不同电离方式,以质子化/去质子化或形成金属离子加合物方式转变为偶电子离子,进一步的解离取决于吸收光波长(光子能量)和强度(光子数量)。提高光程长度,增大有效碰撞截面,使光子与偶电子离子充分作用,这是合理光路设计的关键,直接影响样品准分子离子的解离效率。
5.1 红外多光子解离质谱仪
红外多光子解离技术主要用于离子阱和傅里叶变换离子回旋共振质量分析器。CO2激光常被用作红外多光子解离的光源[94],其输出的 10.6 μm 波长红外激光可引发生物分子中广泛存在的C—C、C—N、C—O、P—O 等化学键振动激活。由于ZnSe对10.6 μm波长红外光吸收很小,而且具有很高的热冲击承受能力,它不仅是高功率CO2激光器的最佳光学材料,也是多光子红外解离质谱仪光学系统的组成部件。
红外多光子解离技术(IRMPD)可以与三维和二维离子阱(线性离子阱)联用[95],它具有比碰撞活化解离(CAD)更加显著的三个优势。第一,在常规CAD分析中,为保证母离子碰撞活化所需能量,通常需要较高射频电压,但是这样会降低对低质-荷比离子的测定能力,虽然可以采取尽可能低的射频电压来捕获离子,仍然无法避免牺牲这些低质-荷比(m/z)碎片离子。而IRMPD与射频电压无关,光解离技术可产生在CAD中不能观察到的碎片离子。第二,在离子阱中使用CAD技术时,只有一个辅助电压激励母离子共振,导致与母离子具有不同共振频率的碎片离子不能被活化。而IRMPD是不依赖于离子共振的解离方式,其母离子和碎片离子都会吸收光子,引发次级离子解离,可得到比CAD更加丰富的碎片离子。第三,IRMPD不是碰撞活化引发的解离方式,不会引发平动激活,有效减少了在使用CAD时,因粒子散射或离子轨迹不稳定所造成的离子丢失。红外激光通过ZnSe小窗后[96,97],由环形电极或端盖电极的小孔进入离子捕集室,辐射时间通常在2 ms到几秒之间,主要由激光功率、光束直径、解离程度、离子阱压力以及目标离子的红外吸收能力等决定。在IRMPD技术中,由于红外光子能量低,样品离子需要经过一段时间(ms级)吸收多个光子才能被激活,在光辐射期间,需要没有离子碰撞的环境以避免离子失活。然而离子阱内常常存在 10-3 Torr 氦气,利用氦气与离子的碰撞消耗离子能量,从而达到阻尼离子运动的目的,这是离子阱技术将离子捕获于阱中心的常用策略。在这种情况下,如何提高光解离效率成为IRMPD需解决的关键问题,现有仪器主要通过增加光程[86]、提高激光功率[98]、热辅助[99]或通过降低压力来减少碰撞失活[100]等技术来提高光解离效率。此外,碎片离子过小是IRMPD面临的另一个问题。由于红外多光子解离没有离子选择功能,分子离子解离后的产物离子会继续吸收光子,裂解反应不断进行,导致某些碎片离子过小,给谱图分析和分子结构解析带来困难。Payne等发展的轴向扩张方法利用辅助射频共振激发产物离子,使他们在z 轴方向振幅增大,引导这些产物离子运动到离子阱边缘,使母离子保留在激光路径中吸收激光能量并发生裂解反应[99]。
红外多光子解离技术可高效地应用于傅里叶变换离子回旋共振(Fourier Transform Ion Cyclotron Resonance, FTICR)质谱[100]。FTICR质谱由加拿大英属哥伦比亚大学(University of British Columbia)的Marshall 和Comisarow于 1974年首次报道于Chemical Physics Letters[101]。离子回旋运动是FTICR质谱分析的重要实验基础,其运动方程式ωc=qB/m(其中ωc为离子回旋运动角频率,B为磁场强度,q为离子电量,m为离子质量)表明离子回旋运动角频率只与离子荷质比 q/m和外加磁场强度B有关。如果使用直线加速器,粒子沿着近于直线的轨道被逐级加速,直线距离越长,离子被加速所获得的动能越高。1930年[102]美国加州大学伯克利分校物理学教授劳伦斯(Ernest Lawrence)提出的回旋加速器理论大大提高了加速器效率,利用较小电场即可获得高达1 MeV的离子动能[103],劳伦斯也因在回旋加速器方面的突出成就获得 1939年诺贝尔物理学奖。基于回旋加速器原理,当施加横向交变电场的频率与离子回旋的频率相等时,离子可被激励到更大轨道半径。FTICR质谱中用于捕获离子的潘宁阱(Penning Trap)具有优越的离子操控性能,它使用均匀轴向磁场和不均匀四极电场束缚离子,即在激发板引入包括所有离子回旋频率的宽频域射频信号(chirp),使所有离子都被共振激发到较大半径做回旋运动,其在接收板上感应产生的镜像电流包含了所有离子信息,该时域信号经傅里叶变换可转化成频域信号,得到以质-荷比(m/z)为横坐标而信号强度为纵坐标的质谱图。与线性离子阱相比,潘宁阱有很高真空度和更长的离子捕获时间,能有效减少碰撞冷却,实现连续多光子吸收和分步激励,从而能增强 IRMPD 效率,提高样品检测灵敏度[104⇓⇓~107]。不仅如此,在红外多光子解离技术中,样品分子离子连续吸收红外光子,不断进行的裂解反应产生大量碎片离子。其他质量分析器受限于质量分辨率,难以对相近或过小的碎片离子进行结构解析,但FTICR具有的高质量分辨率和准确度使其成为多光子解离技术的最佳搭档。FTICR是一种非常独特的质量分析器,其信号检测不是用离子去撞击由微型电子倍增器集成的多通道板(MCP),而是检测由感应产生并包含所有离子信息的镜像电流,依据频率来测定质量-电荷比,有利于分析多光子解离产物。
5.2 紫外光解离质谱仪
紫外光解离可在波长分别介于 200~400 nm 和10~200 nm的近紫外区和远紫外区进行。相对于红外多光子解离,能量更高的紫外光子(3~8 eV)可使分子发生单光子引发的光解离。近紫外光解离技术比红外多光子解离具有更强的选择性,需要特定发色基团。如果样品缺乏发色团,通过化学衍生方法引入特定基团,可提高检测选择性。真空紫外光解离的选择性较差,因为其更高的光子能量,广泛引起生物分子的电子能级跃迁。紫外光解离主要有两种作用形式,第一种可能的途径是分子离子吸收紫外光子后跃迁到高能电子激发态,引发与低能碰撞活化完全不同的独特UVPD裂解途径[110]。也可能弛豫到低电子能态或基态,发生与常规碰撞激活相似的化学键断裂[111]。第二种可能的途径是吸收紫外光子后发生电子脱离[112],电子从多电荷阴离子脱离,成为阴离子自由基,随后与CAD联用引发裂解反应,这种方法也被称为活化电子脱离解离(Activated-Electron-Photo Detachment Dissociation (a-EPD)。
UVPD技术一般采用高能量短脉冲激光(大约几个纳秒),其脉冲数量、能量密度和波长是控制能量转移并影响光激活和裂解效率的关键技术参数。UVPD技术一般采用准分子激光源,包括F2(157 nm,7.9 eV)、ArF(193 nm,6.4 eV)、XeF(351 nm,3.5 eV)以及Nd3+:YAG 固体激光器 (213 nm,5.8 eV;266 nm,4.7 eV;355 nm,3.5 eV)[113]。除此之外,气体放电灯、同步辐射和发光二极管等也可用于光解离。光解离与质量分析器的联用需要对仪器进行两个方面的改造,第一是增加光学系统,采用CaF2或石英窗将紫外光引入仪器。第二是增加触发系统,使光辐射与离子检测同步,控制离子解离程度。
光解离技术也可与飞行时间质量分析器联用。具有不同质-荷比的离子在真空无场区内运动一定距离所需的时间不同,飞行时间质量分析器通过时间来测量离子速度并转换为质-荷比。与离子阱质量分析器相比,飞行时间质量分析器与光解离技术联用面临更多技术挑战,主要是因为激光脉冲与飞行时间难以同步。时间的测量往往需要确定一个起始点,但是这样的时间点难以确定,而且离子初始速度和运动方向各不相同。将连续式离子源变成脉冲式离子源是有效解决途径之一,四极杆-飞行时间(Quadrupole-Time of Flight, Q-TOF)串联质量分析器进一步改进了与光解离技术的联用。离子源产生的分子离子首先在四极杆聚焦,进入正交加速区,并在垂直于入射方向的脉冲电压作用下,获得正交方向恒定速度,这样改善了线性飞行质量分析器初速度和方向不可控的缺点。光解离一般发生在进入飞行时间质量分析器前,在离子捕获腔(Trap Cell)进行。近年来还陆续报道了离子淌度分析器与飞行时间和四极杆质量分析器的联用[118],使光解离技术能够进一步用于解析生物分子异构体。
6 生物分子光解离及结构鉴定
复杂样品分子结构鉴定是化学及相关领域包括生物医学、药学、农学和环境学等急需解决的重要问题,发展新型离子活化/解离(Ion Activation and Dissociation)技术一直是质谱最活跃的研究领域。光解离技术通过光激励可使处于激发态的原子/分子丢失电子,发生由电荷或自由基引发的化学键断裂。或者通过激发态能量弛豫,引发振动激活的化学键断裂。光解离技术能够提供与常规CAD不同的碎片离子,对分析复杂生物大分子(蛋白质和核酸)一级结构、蛋白质翻译后修饰的种类和修饰位点以及有机小分子(如药物和代谢物等)等具有重要意义。
6.1 有机小分子结构鉴定
与蛋白质和核酸相比,有机小分子不具有线性结构,不是特定结构单元的有限重复序列,其多样化结构鉴定不仅要求高度准确地分辨和测定碎片离子质-荷比(m/z),还需要能够高度选择性地溯源辨识不同化学键的断裂途径,最终从获得的质谱图准确地推导出分子结构。而分子离子对特定波长光的选择性吸收,可为溯源识别特定官能团及其相关化学键提供重要依据。
常见有机小分子中的C—C、C—N、O—H、N—H、C  O和C—O键均可由红外激光引发化学键振动激活。
O和C—O键均可由红外激光引发化学键振动激活。图5 (A)和(B)分别是与头孢菌素与青霉素两种 β-内酰胺类药物的红外多光子解离机理[117],由于N原子具有孤对电子,因此质子化头孢菌素正电荷定位于如图5 (A)所示的N原子上。分子吸收多个光子能量,激活与正电荷直接相邻的C—N键,并引发C—N键均裂,经由内酰胺开环而得到m/z 158碎片离子。这个碎片离子在质谱图中为基峰,这是因为四元环具有较大环张力,这种结构特点有助于两个C—N键发生均裂,引发内酰胺环断裂。而所生成产物离子所携带的正电荷离域于p-π共轭体系,具有较高稳定性。研究发现,β-内酰胺类药物均能发生此类裂解,其生成的碎片离子成为鉴定此类药物的特征离子。图5 (B)是青霉素与K+阳离子加合物的红外多光子解离机理,其基峰m/z 160碎片离子也由内酰胺环的断裂产生。
O和C—O键均可由红外激光引发化学键振动激活。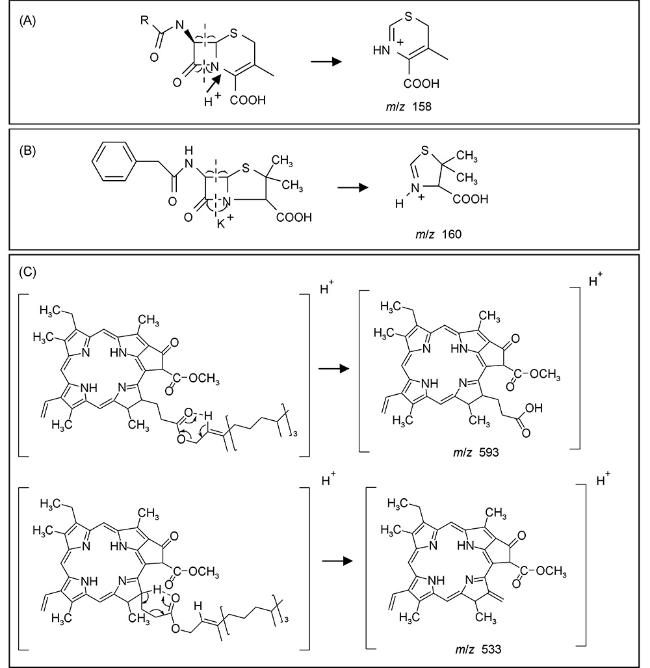
尽管大多数有机小分子化学键都可由红外光子振动激活,但由于每个光子能量低,需要较长时间来积累多个光子能量。紫外光电子具有更高的能量,不同波长紫外光也可由不同化学键选择性吸收,进一步发生由激发态电子引发的化学键断裂,或者先经弛豫至基态,再发生振动激活的化学键断裂。光子能量越高,生成的碎片离子越多,得到的分子结构信息越丰富[119]。
6.2 单糖和多糖结构鉴定
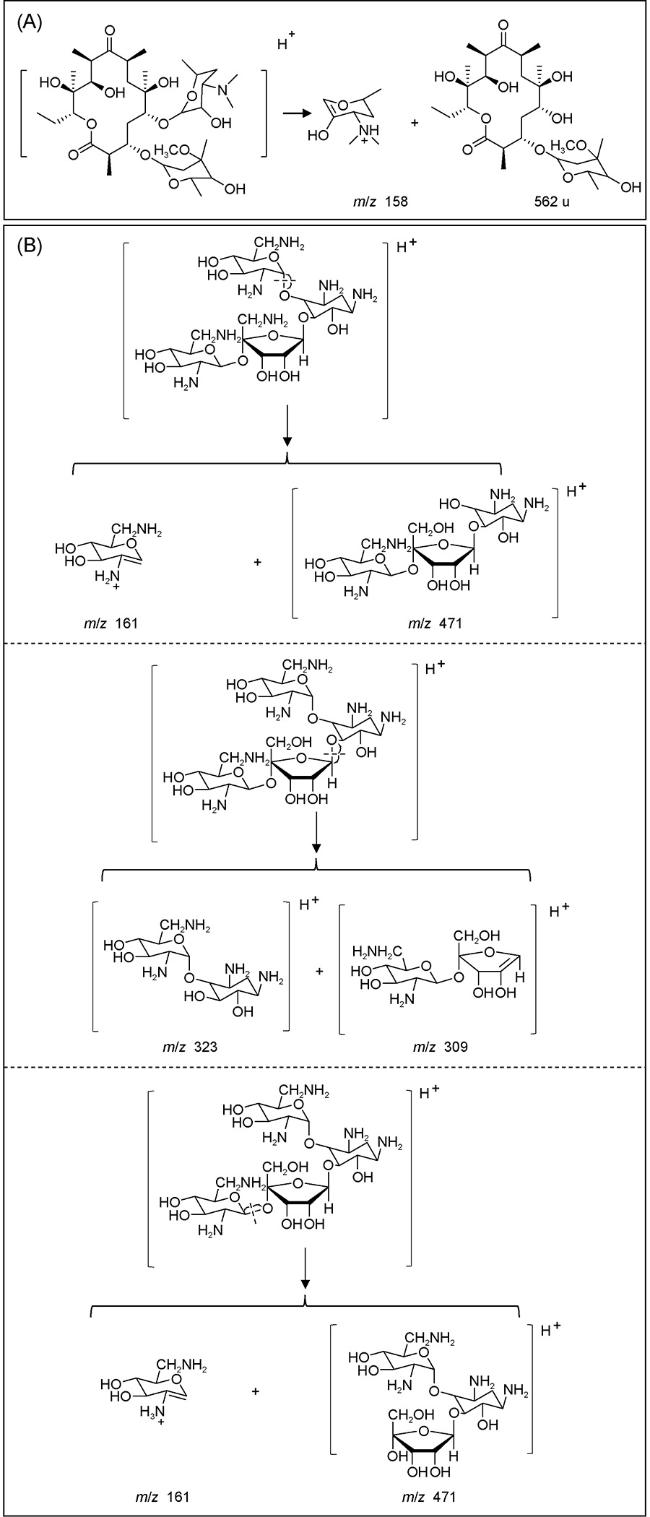
糖是天然产物最大的家族之一,其结构的复杂性不仅在于单糖种类的多样性以及错综复杂的连接方式和分支结构,还在于其常常通过糖苷键与其他分子相连。自然界主要存在四种糖苷,大多数糖苷属于O-糖苷,胺或氮杂环糖基胺化合物为N-糖苷(比如核苷类化合物),而当糖C1直接与配基碳原子相结合时则形成C-糖苷,硫醇和糖的缩合则为S-糖苷。由于缺乏生色团,糖类物质一般在近紫外和可见区没有特征吸收,如果采用近紫外光解离则需要将样品进行衍生化。IRMPD是分析单糖和多糖结构的常用技术[120],CO2激光器所提供的10.6 μm(943 cm-1)红外光子可激活C—O—C振动,但大多数O-糖苷的跨环结构得以保留。图6 (A)和(B)[96]分别表示了红霉素和新霉素通过IRMPD发生C—O—C振动激活的C—O化学键断裂,这两个分子均发生以断裂糖苷链为主的解离,脱去氨基糖苷环。红霉素的大环结构基本不发生裂解,新霉素依次丢失氨基糖苷,可以方便地鉴定糖苷种类。红外多光子解离一般不仅会有糖苷键的断裂,还会引起N-和S-糖苷跨环结构的断裂[121,122],产生丰富的碎片离子,而 CID 需要做数级串联质谱才能达到同样的结果[123]。
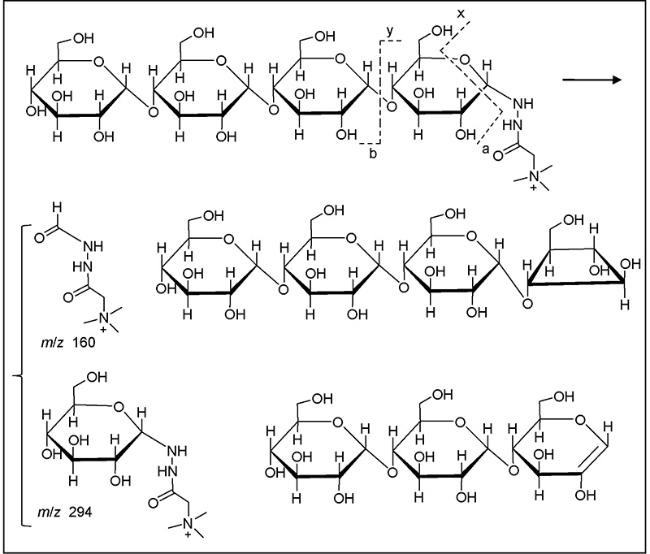
真空紫外光能被大多数糖分子吸收,因为其提供的光子具有较高能量,不仅可激发环外C—O键的断裂,还会引起环内C—O键的断裂。图7 所示为一个寡糖的两种解离途径[124,125],一种是连接两个糖环的糖苷C—O单键解离,形成 y和 b离子,这些离子可提供序列信息。另一种是糖环内C—O键均裂,并引发相邻环内C—C键均裂,最终环结构断裂,生成 x 和 a 离子,这些离子可用于连接方式的鉴定。真空紫外光解离寡糖分子产生的 a、b、y和x离子,与 HCD 得到的碎片离子相似。除了糖苷键和环结构断裂外,也会产生中性丢失碎片。比如去质子化唾酸寡糖在真空紫外光解离下,出现 62 Da 乙二醇中性丢失,为其鉴定提供了特征信息[126]。
6.3 多肽/蛋白质氨基酸序列和翻译后修饰鉴定
IRMPD主要引发多肽/蛋白质C—N 肽键断裂,生成的b离子和y离子是主要碎片离子,这点与CAD非常相似。为确保氨基酸序列和翻译后修饰的准确鉴定,在进行肽键断裂时,需保留翻译后修饰基团信息包括磷酸化、磺酸化、糖基化修饰和二硫键等。由于C—N肽键与翻译后修饰相关的红外吸收频率有较大差异,因此通过控制激发波长有可能实现选择性肽键断裂。比如,P—OH基团的红外吸收频率与CO2激光器提供的943 cm-1光子能量非常相近,因此磷酸化修饰肽会被选择性地引发裂解[127⇓⇓⇓⇓~132]。磺酸化肽的红外多光子解离机理与磷酸化肽非常相似,因为S—OH基团的红外吸收频率也与943 cm-1光子能量相近[133]。IRMPD技术对肽键具有高覆盖断裂能力,减少了CAD技术的离子歧视效应,已用于蛋白质“Top-Down”氨基酸序列分析[134⇓~136]。
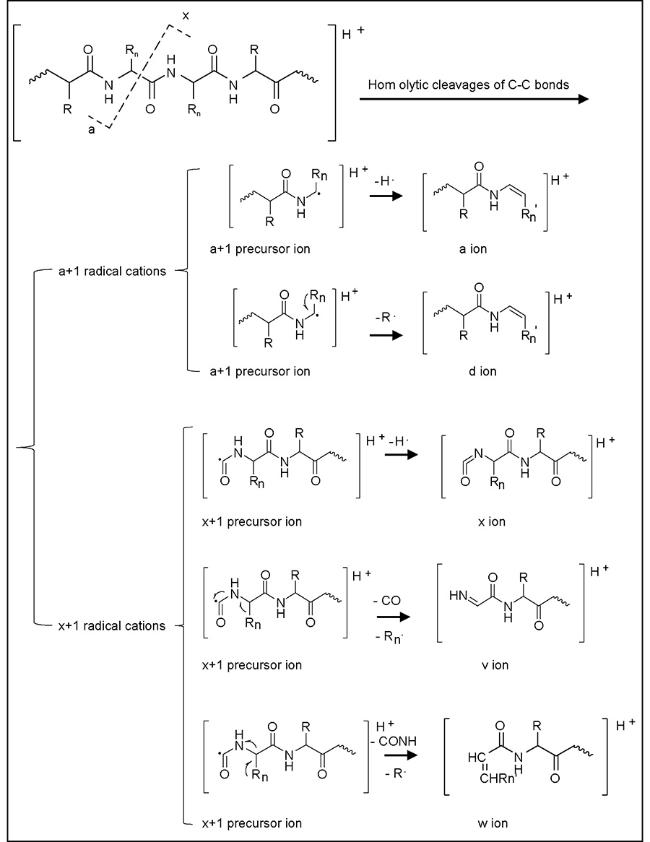
真空紫外光解离技术最早报道于傅里叶变换离子回旋共振质谱仪[145⇓⇓⇓⇓~150],可与MALDI[151⇓⇓⇓~155]和ESI[156⇓⇓⇓⇓⇓⇓⇓~164]等软电离技术相联用。由MALDI产生的准分子离子,其解离是激光热效应和紫外光子共同作用的结果。而由ESI产生的准分子离子被称为“冷”离子[165,166],具有更低的内能,即使与离子阱中缓冲气体碰撞也难以发生振动激活化学键断裂,这样就确保了离子从形成到进入质量分析器之间的几毫秒时间段内发生由光电子引发的特异性化学键断裂。图8 显示了157 nm真空紫外光电离不同离子的机理[167],C—Cα 键均裂产生a+1和x+1阳离子自由基,再经自由基中心引发的邻位化学键均裂,并消除H原子,分别生成偶电子离子a 和x,而经侧链丢失则分别产生d、v和w离子, 较少产生b、y和c、z型离子。生成 x 离子还是a离子取决于质子化碱性氨基酸在多肽中的位置,如果碱性精氨酸出现在N端,电荷相应就会保留在 N 端,生成a离子。如果碱性精氨酸出现在C端,碎片离子电荷保留在 C 端,主要产生x离子,这种现象被称为“精氨酸效应”。与此相反,赖氨酸在C端主要产生y离子和 b离子,而将赖氨酸侧链氨基胍基化,则会发生与精氨酸类似情况,因此紫外光电离技术可为识别C端精氨酸肽段提供相关信息[168]。产物离子的形成与化学键断裂所需的能量和产物稳定性有关,比如天冬氨酸残基消除发生于侧链,而谷氨酸残基消除则发生于主链,这是因为天冬氨酸残基形成的 Cα Cβ 键能够与酰胺键共轭,结构更加稳定。一般情况下,d 离子不会出现在无 β—C 取代的甘氨酸和丙氨酸残基中,而消除芳香环需要更高能量,所以芳香氨基酸残基也不会产生d离子[165]。当利用紫外光去解离ECD和ETD产生的气态离子时,如果精氨酸在N端,则主要产生a离子,符合精氨酸效应。但如果精氨酸处于C端,则会断裂N-Cα键形成c和z离子[169]。离子解离也与电荷定位有关,如H+和Na+定位于碱性氨基酸残基,容易发生电荷引发的化学键断裂[170]。
Cβ 键能够与酰胺键共轭,结构更加稳定。一般情况下,d 离子不会出现在无 β—C 取代的甘氨酸和丙氨酸残基中,而消除芳香环需要更高能量,所以芳香氨基酸残基也不会产生d离子[165]。当利用紫外光去解离ECD和ETD产生的气态离子时,如果精氨酸在N端,则主要产生a离子,符合精氨酸效应。但如果精氨酸处于C端,则会断裂N-Cα键形成c和z离子[169]。离子解离也与电荷定位有关,如H+和Na+定位于碱性氨基酸残基,容易发生电荷引发的化学键断裂[170]。利用真空紫外光解离技术鉴定翻译后修饰,可以较好地保留修饰位点[171]。研究发现,磷酸分子中性丢失与光照时间密切相关。当用157 nm 真空紫外光解离丝氨酸和苏氨酸磷酸化肽时,如果控制激发时间在1 μs内,产物离子几乎没有98 Da磷酸中性丢失。超过1 μs即开始发生磷酸基团丢失,到13 μs 时离子普遍丢失了磷酸基团。相比之下,与酪氨酸相连的磷酸基团比丝氨酸和苏氨酸更稳定,很少有产物离子丢失磷酸基团,即使光激发时间到13 μs 也没有观察到磷酸基团的丢失[172]。由于磷酸化肽携带负电荷,在负离子模式下解离效率更高,可形成大量的a和x离子,并且将磷酸基团较完好地保留在碎片离子中,便于鉴定修饰位点[173]。
6.4 核酸序列分析
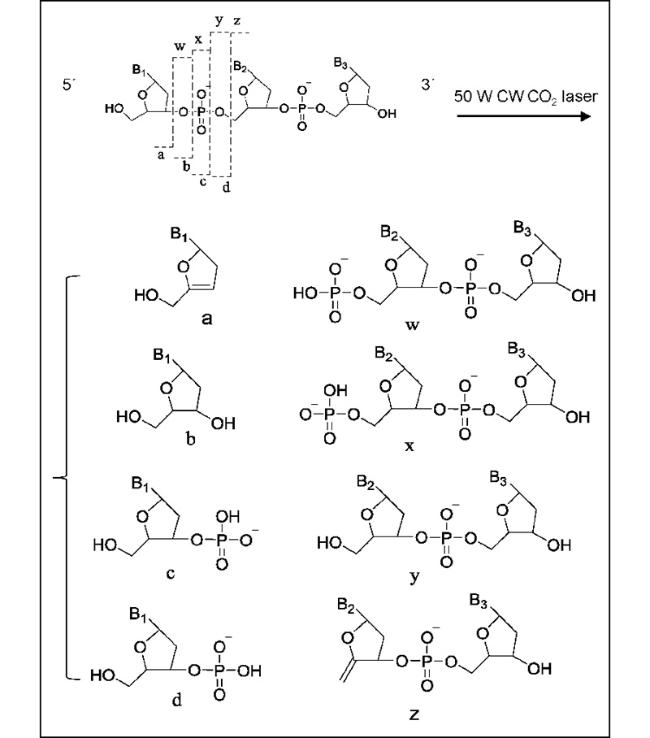
将质谱用于核酸分析具有较大挑战性,因为核酸的磷酸酯带负电荷,而质谱负离子模式检测灵敏度一般比正离子模式低。其次,核酸只有四个碱基,解离后产生大量同分异构碎片离子,给序列分析带来一定困难。过去主要利用CAD解离技术获得碎片离子,为了获得更加丰富的碎片离子,各种光解离技术如IRMPD和UVPD逐渐发展起来。由于核苷酸中磷酸基团对常用CO2激光器发射的红外光子(10.6 μm波长)具有较大吸收截面[174],在低至微秒级时间内,核酸双链结构即可解离为单链,再连续吸收多个红外光子即可进一步引发化学键断裂,对寡聚核苷酸、miRNA和siRNA等均能获得较完整序列信息[174,175]。如图9 所示,IRMPD通过激励P—O和C—O键,积累多光子能量引发振动激活的化学键断裂,生成包含序列和修饰基团信息的碎片离子。离子解离程度与光子辐射时间有关,改变激光强度和辐射时间均可改变碎片离子组成。但辐照时间过长,可导致两个5’ P—O键断裂,生成干扰序列分析的内离子(internal ion),以及w离子和丢失碱基的a离子。此外,CAD技术不能获得的小质子化碱基离子,包括[GH+]、[AH+]以及[CH+]等,也可通过IRMPD解离技术获得。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
UVPD与IRMPD原理截然不同,它主要通过电子的光脱离(Electron Photodetachment, EPD)进行核酸解离,即去质子化核酸吸收高能量紫外光子后,失去电子成为奇电子离子。这种奇电子离子内能不高,不会自发解离成碎片离子,需与CAD碰撞活化联用以获得碎片离子,这个过程被称为活化-电子光脱离(Activation-Electron Photodetachment, a-EPD)。其奇电子离子经碰撞活化后裂解,可获得完整a、b、c、d、x、y和z系列离子以及低质量w1、z1和a1离子,减少了碱基丢失,提高了核酸序列的覆盖度。
7 结论与展望
生物分子复杂多样的结构是执行其独特功能的分子基础,其复杂结构的准确鉴定要求发展多样化分析方法以揭示不同的结构特点。电离/解离是质谱分析的关键,不同中性样品分子经电离转变为气态带电荷离子,其中的偶电子离子具有较高稳定性,还需通过其他解离技术进一步解离为碎片离子。光电离/解离技术基于不同化学键或官能团对特定波长光子的吸收,与其他解离技术比如碰撞活化解离相比,光电离/解离技术提高了选择性和解离效率。从小分子结构到大分子序列、共价修饰以及结合位点,从遗传信息储存者核酸到功能执行者蛋白质以及药物、代谢物等领域,光电离/解离技术都发挥了重要作用(表2 )。目前光电离/解离技术还存在一些技术挑战,限制了其在生命科学领域更加广泛的应用。由于现有光电离/解离技术一般是使用单一波长激光光源,不能进行连续波长扫描,因此只能得到由特定化学键断裂形成的碎片离子。生物分子结构多样,存在大量异构体,进一步将光谱和质谱联用,不仅可获得准确的质量-电荷比,还能得到官能团信息,预期可进一步提高对未知分子的结构鉴定能力。
表2 通过光电离和解离对生物分子的质谱鉴定Table 2 Mass spectrometric identification of biological molecules with photo ionization and dissociation |
| Samples | IR1 | UV2 | ||||
|---|---|---|---|---|---|---|
| Principles | Features | Disadvantages | Principles | Features | Disadvantages | |
| Small organic molecules | Bond cleavages by vibrational excitation | Selective absorption in IR region by different bonds | Need multiple IR photons to activate bond cleavages due to the low energy of each IR photon | Electron/charge-directed bond cleavages + vibration excited bond cleavages | Do not need multiple UV photons | Extensive bond cleavages/Need chemical derivatization in the near UV light region |
| Monosaccharides/polysaccharides | Distinguish O-, N-, C- and S- glycosidic bonds | ·Characteristic a, b, y and x ions as well as neutral losses in the vacuum UV region. | ||||
| Peptides/Proteins | ·Characteristic b and y ions resulting from C-N bond cleavages. ·Identification of fragile posttranslational modification that is difficult in CAD3. | ·Selective cleavages of S-S bonds in the near UV region. ·Characteristic a, x, d, v and w ions in the vacuum UV region. ·Arginine effects on the production of a or x ions | ||||
1Infrared (IR),2Ultraviolet (UV),3Collision activated dissociation (CAD) |