




1 引言
N,N-二甲基甲酰胺(DMF)不仅是一种重要的“万能溶剂”,同时作为医药、农药和有机合成的重要中间体被广泛应用于工业生产[4]。目前,工业上DMF的合成方法主要是通过甲醇钠催化二甲胺和CO直接反应生成DMF[5]。此生产方法具有原料来源广泛、适合大规模连续生产等优点,美国、日本以及我国上规模企业等大多采用此法。但是此方法在实际生产中,会发生一些副反应,产生一甲基甲酰胺(MMF)、二甲基乙酰胺(DMAC)、甲酸以及盐类物质氢氧化钠、甲酸钠、碳酸氢钠、碳酸钠等,因此每隔一段时间需要停产进行固体沉积物的清除及设备保养。而且,该方法依赖于不可再生的煤炭资源,在DMF的大规模生产中消耗量大,不利于可持续发展[6]。
因此,以CO2作为羰源,通过高效催化体系的构建实现二甲胺合成DMF无疑是一条绿色、可持续的途径。本文综述了近年来二甲胺与CO2反应合成DMF的反应研究及发展现状。在此基础上,对不同催化体系的典型反应机理进行了讨论,并对未来可能的发展方向进行了探讨和展望。
2 H2作为还原剂
2.1 均相催化体系
2.1.1 贵金属催化体系
1970年,Haynes[7]首次报道了均相催化二甲胺、CO2和H2制备DMF(图1 )。金属配合物包括CoH(dppe)2、(PPh3)3RhCl、(PPh3)3CuCl、(PPh3)3RuCl都具有很高的催化活性,其中(PPh3)2(CO)IrCl活性最好,在125 ℃、总压力为54 bar ( = )下,DMF的TON达到1200。该工作开启了CO2甲酰化合成DMF的先河。
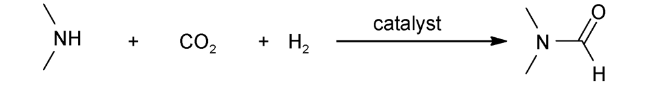
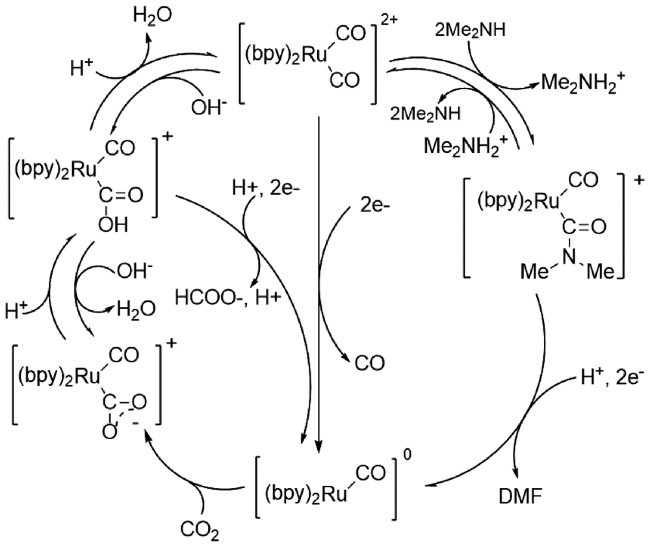
1987年,Tanaka等[12]首次报道[Ru(bpy)2(CO)2]2+(bpy=2,2-二吡啶)电催化二甲胺与CO2合成DMF。经傅里叶变换红外光谱(FTIR)和核磁共振氢谱(1H NMR)研究发现该催化剂在二甲胺存在时可快速和胺结合生成[Ru(bpy)2(CO)C(O)NMe2]+,经过双电子还原生成DMF和五配位的[Ru(bpy)2(CO)]0,这个零价Ru物种将再度吸收CO2,使得反应持续进行(图3 )。
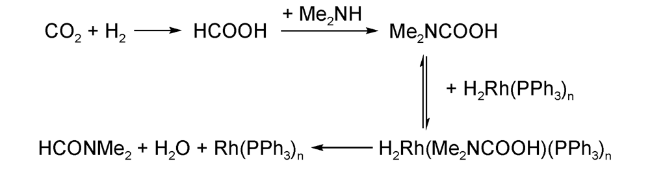
二甲基铵·二甲基(DIMCARB)在60 ℃以上分解成二甲胺和CO2,常被用作二甲胺的来源。1994年,Noyori等[13]以DIMCARB作为二甲胺的前驱体,在超临界反应条件下(130 ℃, 13 MPa CO2, 8 MPa H2),RuCl2[P(CH3)3]4催化合成DMF的TON高达370 000,TOF高达10 000 h-1。这主要是由于在超临界条件下,整个反应处于均相状态,H2、CO2的溶解度以及反应速率均得到了大幅提升。在放大反应过程中,通过周期性补充H2和scCO2保持scCO2压力与氢胺比,在100 ℃条件下DMF的TON最高可达到420 000[14]。受Noyori的工作启发,Baiker等[15]制备了更高活性的[RuCl2(dppe)2]配合物,在无须任何溶剂、100 ℃、13 MPa CO2和8.5 MPa H2条件下,DMF的TON达到740 000,TOF高达360 000 h-1。通过放大反应,1 g Ru催化剂在2 h内可以生产530 kg的DMF。
对于在超临界流体中的化学反应,反应结果受催化体系相态影响很大,即使在反应的初始阶段,包括分子催化剂、起始原料和溶剂在内的所有组分都是可溶性非超临界流体,反应过程中各组分经常发生相分离导致反应效率降低。2001年,Ikariya等[16]根据CO2甲酰化反应制备了含三(羟甲基)膦的水溶性Ru(Ⅱ)配合物RuCl2[PH(CH2OH)2]2[P(CH2OH)3]2,采用这种水溶性三烷基膦配体可以克服反应过程中因相分离而导致的催化剂失活问题,在scCO2-H2O两相体系,CO2合成DMF反应中TON可达到10 000。此后,Behr等[17]制备出了Ru-膦络合物用于催化CO2甲酰化反应,并应用ICP测试催化剂在反应过程中的流失量。配合物[RuCl2(dppb)2]在CO2甲酰化合成DMF反应中的TON最高为4100,催化剂流失量最低为2.9%。
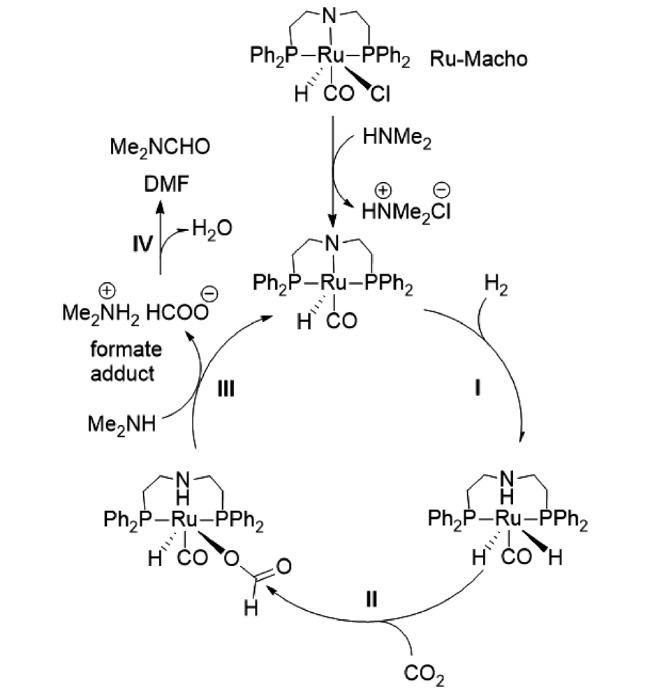
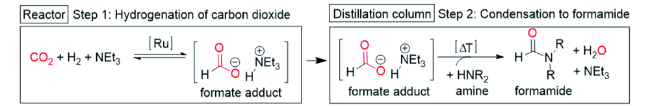
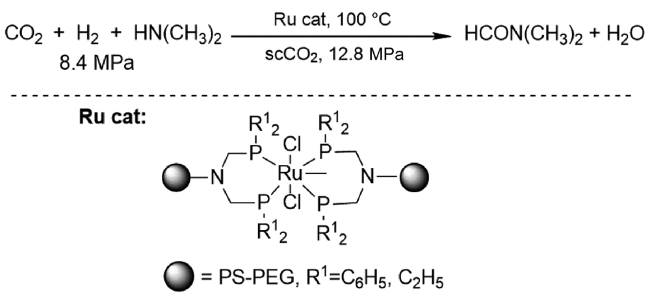
2017年,Vorholt课题组[21]报道了RuCl3·H2O和2,2'-二[(二苯基膦基)甲基]-1,1'-联苯(BISBI)原位生成Ru配合物催化二甲胺合成DMF反应。采用 2-乙基己醇为两相溶剂,催化剂被固定在非极性溶剂中,产物DMF被原位萃取至水相。催化剂重复10次平均收率为31%。随后,他们由釜式设备转移至连续生产的小型工厂,DMF收率仍可保持在31%,TON达到4942,所得产物通过蒸馏分离得到纯品[22]。通过机理研究(图5 ),发现CO2合成DMF的中间产物是甲酸盐而不是CO,CO2和胺的相互作用是影响产物收率的主要因素[23]。为了改善甲酸酯中间体与胺,特别是与非碱性芳香胺的缩合反应速率,该课题组开发了一种两步法工艺来改进缩合和后续产品分离(图6 )。即在小型反应器装置中将CO2加氢生成甲酸盐,然后在蒸馏纯化过程中与胺缩合。新一代甲酰胺合成工艺可快速形成甲酸盐,进一步选择性缩合生成所需的甲酸铵[24]。
2.1.2 非贵金属催化体系
由于贵金属在地壳中的含量稀少,使用成本较高,因此,开发高效、温和的非贵金属催化体系替代贵金属体系具有重要的现实意义。
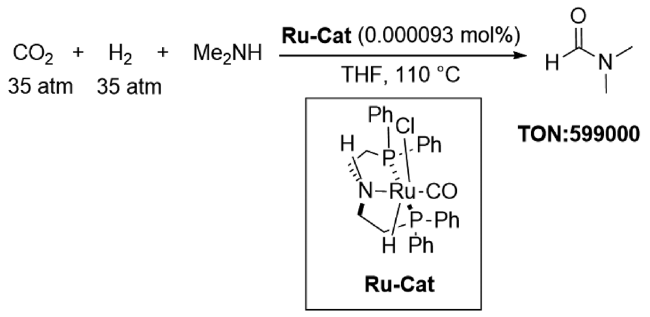
2001年,Takashi课题组[25]通过在甲苯中回流[MoH4(dppe)2]和PhSiH3得到MoH3{Si(Ph)[Ph2PCH2CH2P(Ph)C6H4-o]2}](图7 ,R1),并首次将其应用于CO2合成DMF。作者认为,在110 ℃、25 atm CO2、35 atm H2条件下,CO2插入Mo—H键形成的活性中间物种是生成DMF的活性催化剂和关键中间体。
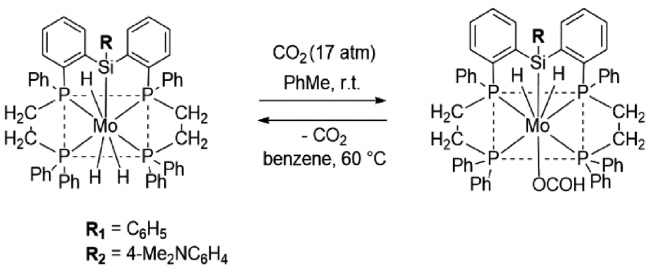
如果将上述催化剂中的R官能团替换为给电子官能团,其中,当R替换为4-Me2NC6H4(R2),二甲胺甲酰化反应TON由92提升至130。这可能是由于给电子官能团可以提升Si原子的电子密度[26]。
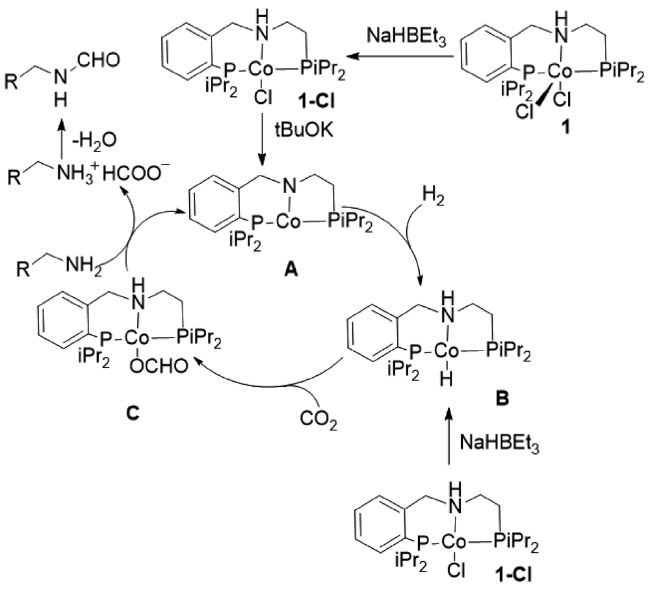
2017年,Milstein等[30]用Co pincer配合物催化剂,以NaHBEt3和BuOK为助剂,在CO2/H2各30 bar、150 ℃下,二甲胺合成DMF收率为99%。机理如下(图8 ):Co pincer配合物分别与NaHBEt3和BuOK作用生成不饱和中间体A,随后加氢生成中间产物B,CO2插入Co—H形成中间体C,过量胺与C去质子化、脱水形成甲酰胺。
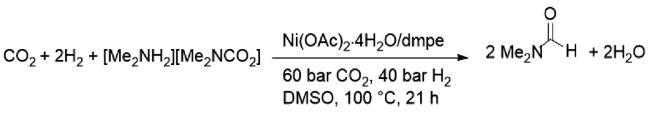
同年,Jessop等[31]开发了一种非贵金属Ni催化CO2胺化制备甲酰胺的方法。其中,Ni(OAc)2·4H2O与1,2-双(二甲基瞵)乙烷(dmpe)原位配位形成的Ni-膦体系催化活性最高(图9 )。在100 ℃、60 bar CO2和40 bar H2条件下反应21 h,TON为632。
随后,他们对Ni配合物催化剂进行优化,制备了cis-[NiCl2(dmpe)],该催化剂配位更不饱和,更容易活化CO2,因此在反应过程中催化活性更高。[NiCl2(dmpe)]催化CO2合成DMF反应TON最高可达6300[32]。
2.2 多相催化体系
2.2.1 贵金属催化体系
2003年,Ikariya等[37]将Ru(Ⅱ)配合物键合在两亲性树脂(PS-PEG)上,其两亲性结合了均相催化剂亲水性和多相催化剂可重复利用的优点,以scCO2作为溶剂及羰源,二甲胺与H2反应生成DMF,在100 ℃下反应15 h,TON为1960,该催化剂可重复使用5次,催化活性无明显下降(图10 )。
2020年,Yoon等[38]将RuCl3与膦基多孔有机聚合物配位形成Ru@PP-POP催化剂用于CO2合成DMF反应,初始TOF为29 000 h-1,而TON则高达160 000(图11 )。该催化剂在加氢过程中表现出良好的选择性和稳定性,已成功应用于连续流反应器,具有一定的工业化规模生产前景。
2014年,石峰课题组[41]以贵金属Pd作为活性中心催化胺甲酰化反应,他们将Pd还原沉积在水热法合成的Al2O3纳米棒(Al2O3-NR)上,制备了一种简单高效的Pd/Al2O3-NR-RD催化剂。在1 MPa CO2、2 MPa H2、130 ℃条件下,该催化剂在催化胺甲酰化反应中表现出很高的活性,其中由二甲胺合成DMF的分离收率为84%。随后,他们通过原位还原Pd(NH3)xCly/C生成一种羟基功能化碳材料负载的高活性纳米Pd/C催化剂。其中,羟基可以有效地调节Pd/C的催化活性,也可以调节碳表面的亲/疏水性,促进Pd位点周围的CO2和胺吸附。在合成DMF中,在1 MPa CO2,2 MPa H2,105 ℃条件下反应24 h,DMF的分离收率为53%[42]。2019年,该课题组再次创新性地将Pd担载在凹凸棒(PAL)上制备了Pd/PAL催化剂,利用凹凸棒丰富的沸石状通道、高比表面积、具有多种类型的酸和碱的活性位点的优势,Pd/PAL在96 ℃、1 MPa CO2、2 MPa H2条件下,在二甲胺合成DMF反应中收率达到86%[43]。
复旦大学曹勇课题组[44]报道了一种高效的TiO2负载Ir催化剂Ir/HSA-TiO2-A,在140 ℃、无有机溶剂条件下直接由CO2、H2和NHMe2水溶液合成DMF,收率达到93.5%。
2016年,Jain等[45]报道了一种氧化石墨烯(GO)接枝Ir配合物催化剂GO-Ir,在总压力为60 bar( / =1)、100 ℃条件下,由40 wt%的NHMe2水溶液催化合成DMF收率为92%,选择性为95%。
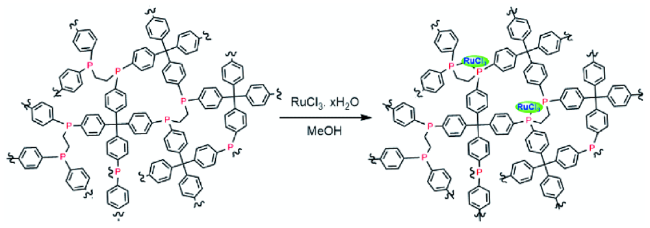
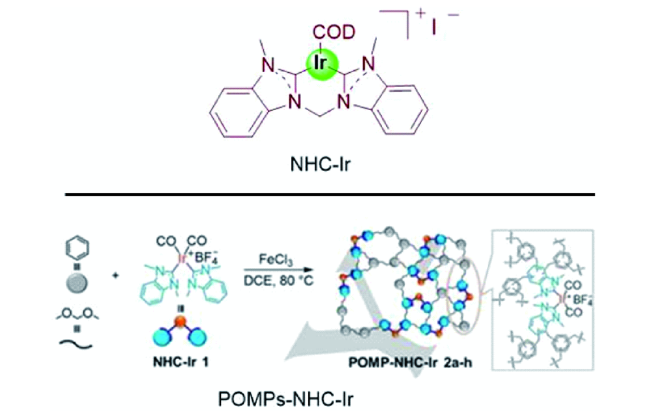
随后,涂涛等[46]首次使用氮杂卡宾(NHC)配位Ir生成NHC-Ir,这种高效固体分子催化剂可以重复使用10次而不发生明显失活。在总压力为60 bar( / =1)、100 ℃条件下,高效催化各类胺甲酰化反应中,其中由DIMCARB合成DMF的分离收率为53%。2021年,他们用直接超交联方法制备多孔有机聚合物POMPs-NHC-Ir(图12 )。该催化剂具有性质稳定、CO2吸附强、多级孔道分布、金属中心高度分散等优点。在120 ℃、无需溶剂、碱和添加剂条件下,采用ppm级的催化剂,在DMF合成中获得了创纪录的TON (1.58×106),固体POMP-NHC-Ir催化剂可以重复使用12次而没有明显失活,突出了其良好的工业应用潜力[47]。
2.2.2 非贵金属催化体系
2010年,韩布兴课题组[50]以DIMCARB为胺源,针对CO2加氢生成DMF反应制备了一系列Cu多相催化剂,经过活性测试,发现Cu/ZnO(Cu∶Zn =3∶2)的催化活性最高,在140 ℃条件下反应6 h,收率可达97%。该体系中,Cu与ZnO具有很好的协同催化效果,开辟了非贵金属多相催化合成DMF的先河。
此后,Jain等[51]在回流条件下,将甘氨酸和CuCl2混合在乙醇中制得Cu(gly)2配合物,使用该催化剂在无溶剂条件下,85 ℃、4 h内DMF的收率高达91%。
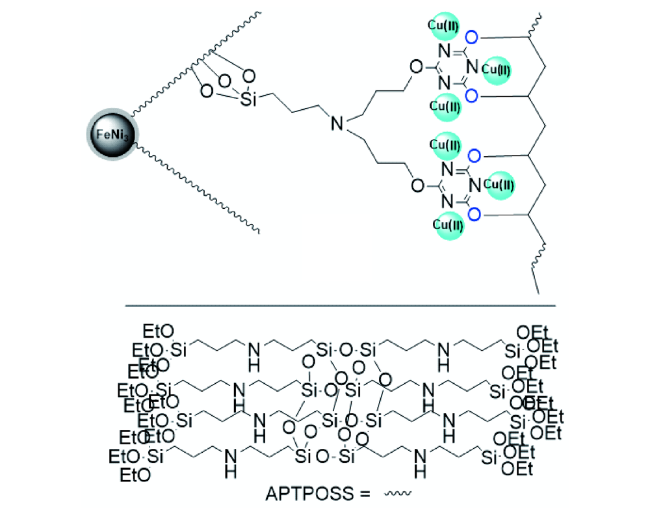
2018年,Seyed Mohsen等[52]报道出将非贵金属铁配合物固载在纳米金属颗粒表面,制备配合物FeNi3/KCC-1/APTPOSS/TCT/PVA/Cu(Ⅱ) MNPs(图13 )。当CO2为1.5 MPa、H2为2 MPa时,在1,4-二氧六环中回流80 min,DMF的收率为91%。
石峰等[53]在共沉淀法制备的CuAlOx多相催化剂表面原位沉积氮掺杂碳膜,将其应用于CO2选择性合成DMF反应。经DFT计算发现,CuAlOx表面原位生成的层状碳膜可以抑制DMF进一步加氢生成三甲胺。重复使用三次以后,在160 ℃、3 MPa CO2、7 MPa H2条件下,反应24 h,DMF的收率可达到97.3%。
2021年,Sun与Zhiani等[54]首次将介孔Sn(Ⅳ)掺杂DFNS负载的BaMnO3纳米颗粒(BaMnO3/SnD NPs)作为催化剂用于CO2甲酰化反应中,在100 ℃下,总压力为5.0 MPa(CO2∶H2=1∶1)条件下,DMF收率为92%。
3 其他还原剂
除了H2以外,其他高活性还原剂如硅烷(R3Si-H)、硼烷(R2B-H),也被应用于CO2加氢反应中。相比于H—H键,Si—H与B—H键能更低,因此更易被活化,反应条件更温和。
3.1 硅烷作为还原剂
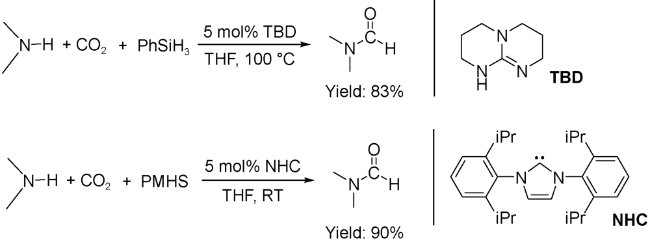
在有机催化过程中,1,5,7-三氮杂双环(TBD)是促进CO2插入N—H键形成CO2加合物的有效催化剂。2012年,Cantat等[55]以TBD催化胺甲酰化反应,在100 ℃、2 bar CO2、无溶剂条件下,以PhSiH3为氢源,DMF的分离收率达到83%。相比于其他氢硅烷,聚甲基氢硅氧烷(PMHS)更稳定,能够长时间地储存而不失活,而且PMHS是硅工业的副产物,是一种廉价易得的还原剂。但是PMHS的反应活性很低。为了活化PMHS,他们采用活性更高的氮杂卡宾(NHC)作为有机催化剂,在室温下,IPr型卡宾化合物催化二甲胺与1 bar CO2的反应收率可以达到90%(图14 )[56,57]。
2013年,Baba等[58]报道了一种Cu(OAc)2·H2O与配体1,2-双(二苯基膦)苯原位生成的铜-二膦配合物催化剂在1 atm CO2、80 ℃条件下以PMHS作为氢源催化胺甲酰化反应,其中,DMF收率达到75%。
N-杂环烯烃(NHO)是一类特殊的新型有机配体,具有很强的供电子特性。Bhanage等[61]以PMHS或9-BBN作为还原剂,以NHO有机催化剂促进胺的N-甲酰化反应,在80 ℃、2 MPa CO2条件下DMF的收率分别为72%或78%。反应过程中原位生成的两性离子NHO-羧酸盐(NHO-CO2)是CO2活化的关键。
3.2 硼烷作为还原剂
3.3 其他还原剂
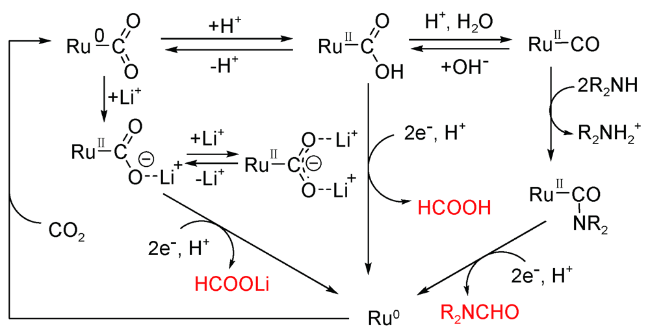
2014年,Tanaka课题组[67]开发了[Ru(bpy)2(CO)2]2+光催化CO2选择性合成DMF反应体系。经过机理研究表明,Me2NH先亲核攻击[Ru(bpy)2(CO)2]2+形成了前驱体[Ru(bpy)2(CO)(CONMe2)]+,进而转化为DMF。在该过程中,Me2NH既是电子供体又是反应底物,而$Me_2 NH_2^-$提供质子。另外,由于Li+形成的Ru-CO2支架结构具有强稳定性,阻止了Ru-CO络合物的形成。因此,Li+的存在可以极大地抑制DMF的产生(图15 )。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
4 结论与展望
CO2既是一种温室气体,又是相对安全、清洁、低成本的原料。DMF用途广泛,市场潜力巨大。因此,由CO2合成DMF将变废为宝,实现CO2的高值化利用。本综述按照不同氢源和催化体系对CO2与二甲胺合成DMF的研究进展进行了分类介绍。该领域已经取得了较好的研究进展,例如:催化体系已经涵盖了热催化、光催化和电催化领域;活性催化剂包括贵金属、非贵金属、分子筛和有机催化剂;催化效率如TON也达到了百万级,而收率也高达99%。
虽然取得了以上进展,但该领域仍有一些挑战亟待解决。(1)目前已有的高效、温和的催化体系多以硅烷或硼烷作为还原剂,反应过程中会产生当量的硅烷醇和硼酸,不符合原子经济性的要求;而以绿色、清洁、经济的H2作为还原剂时,反应条件较为苛刻,包括反应压力较高、已报道的高TON体系很多需要引入超临界反应体系,对设备要求苛刻。(2)均相催化体系多涉及对空气和水十分敏感、价格昂贵的膦配体的使用,催化剂成本高、合成复杂,使实际应用变得困难;另外,均相催化体系面临着催化剂回收问题。(3)非贵金属催化体系面临着反应条件较为苛刻、催化剂用量高、转化数不高等问题,远远达不到工业化生产的要求。如何构建高效、经济、绿色的非贵金属催化体系,实现温和条件下的DMF合成仍是未来需要重点关注的问题。(4)通过表征技术揭示催化剂(尤其是多相催化剂)构效关系,通过原位光谱技术对催化过程进行机理研究,将为催化剂设计和高效催化体系的建立提供理论参考。总之,利用现代催化剂表征手段和理论计算方法,开发高效、经济、具有工业化应用前景的CO2合成DMF催化反应体系仍将是未来的发展方向。