




1 引言
2 SCR反应机理及催化剂设计
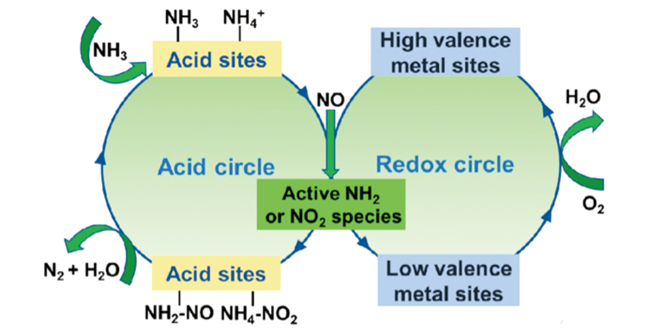
3 Ce基脱硝催化剂
3.1 单一Ce基催化剂
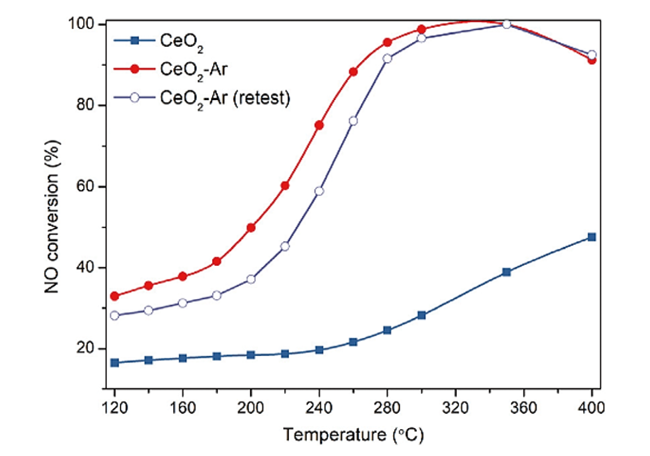
图2 空气中焙烧的CeO2、Ar氛围焙烧的CeO2-Ar和重复测试的CeO2-Ar的NO转化率(反应条件:150 mg催化剂、[NO] = [NH3] = 650 ppm、[O2] = 5 vol%、气流300 mL·min-1)[8]Fig. 2 NO conversion of NH3-SCR over CeO2 calcined in air, CeO2-Ar calcined in Ar and retested CeO2-Ar (reaction condition: 150 mg catalysts, [NO] = [NH3] = 650 ppm, [O2] = 5 vol% and flow rate of 300 mL·min-1)[8]. Copyright 2020, Elsevier |
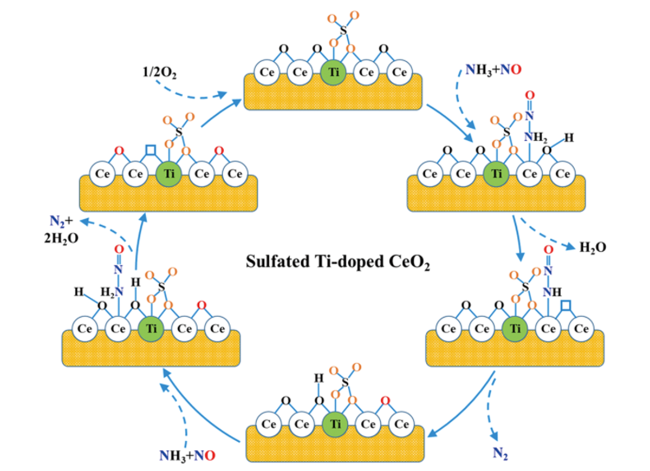
3.2 Mn-Ce基催化剂
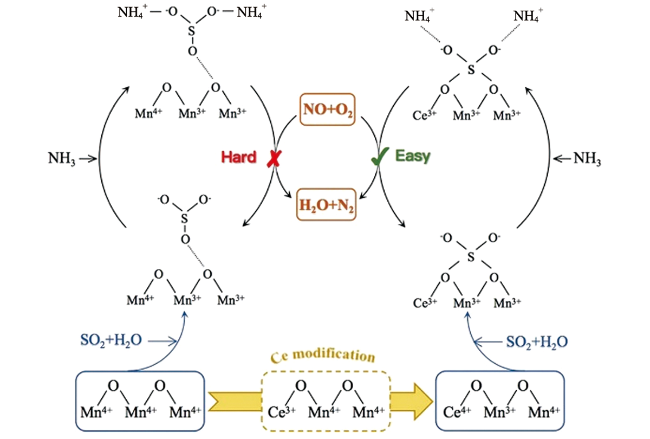

表1 部分Mn-Ce催化剂的脱硝活性列表Table 1 summary of catalytic activities of some Mn-Ce catalysts |
| Catalysts | Preparation | Reaction conditions | NO conversions | ref |
|---|---|---|---|---|
| Mn3CeOx | Coprecipitation | [NO] = [NH3] = 500 ppm, [O2] = 5 vol%, GHSV = 100 000 h-1 | 83%~97% (125~200 ℃) | 22 |
| C -K-OMS-2 | Reflux | [NO] = [NH3] = 600 ppm, [O2] = 6 vol%, GHSV = 64 000 h-1 | 100% (140~230 ℃) | 45 |
| Ce-Mn/ZSM-5 | Impregnation | [NO] = [NH3] = 1000 ppm, [O2] = 3 vol%, GHSV = 22 500 h-1 | 80% (265~465 ℃) | 46 |
| C Mn/TiO2 | in situ growth | [NO] = [NH3] = 1000 mg· , [O2] = 3 vol%, GHSV = 10 500 h-1 | 85%~100% (100~300 ℃) | 47 |
| 5%Mn-Ce/ACN | Impregnation | [NO] = [NH3] = 800 ppm, [O2] = 3 vol%, GHSV = 22 070 h-1 | >90% (120~250 ℃) | 48 |
| Mn-Ce(0.4)/AC | Impregnation | [NO] = [NH3] = 500 ppm, [O2] = 5 vol%, GHSV = 18 000 h-1 | >90% (100~300 ℃) | 49 |
| Mn-Ce/AC | Impregnation | [NO] = [NH3] = 500 ppm, [O2] = 11 vol%, GHSV = 12 000 h-1 | 54%~92% (100~200 ℃) | 50 |
3.3 Fe-Ce基催化剂
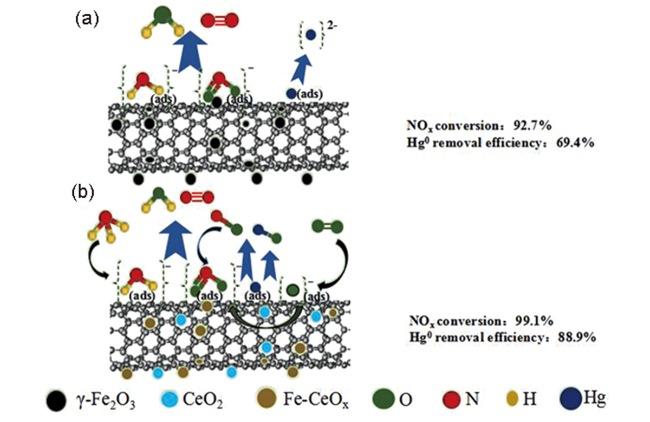
3.4 V-Ce基催化剂
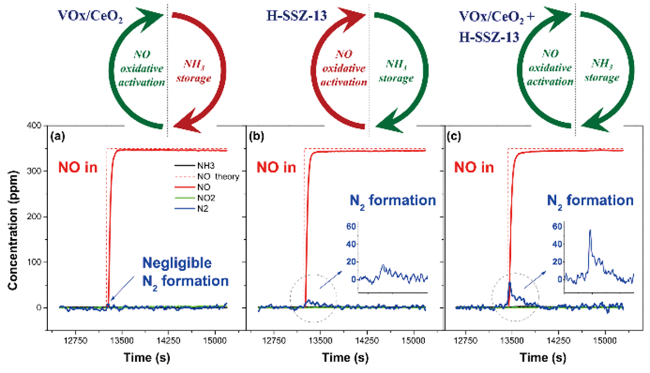
图7 表面酸性和氧化还原性能组合示意图,以及瞬态响应测试结果,(a)VOx/CeO2 (0.3 g),(b)H-SSZ-13 (0.03 g),(c)VOx/CeO2 + H-SSZ-13 (0.3 g + 0.03 g)的机械混合物,T = 473 K, O2 = H2O = 0%, NO = 350 ppm, 流速 = 3.33 cm3·s-1 NTP[26]Fig. 7 Schematization of surface acidity and redox properties and transient-response experiment results: NH3 adsorption (not shown) + transient NO pulse over pretreated (a) VOx/CeO2 (0.3 g), (b) H-SSZ-13 (0.03 g) and (c) mechanical mixture of VOx/CeO2 + H-SSZ-13 (0.3 g + 0.03 g). T = 473 K, O2 = H2O = 0%, NO = 350 ppm, flow rate = 3.33 cm3·s-1 NTP[26]. Copyright 2020, Elsevier |

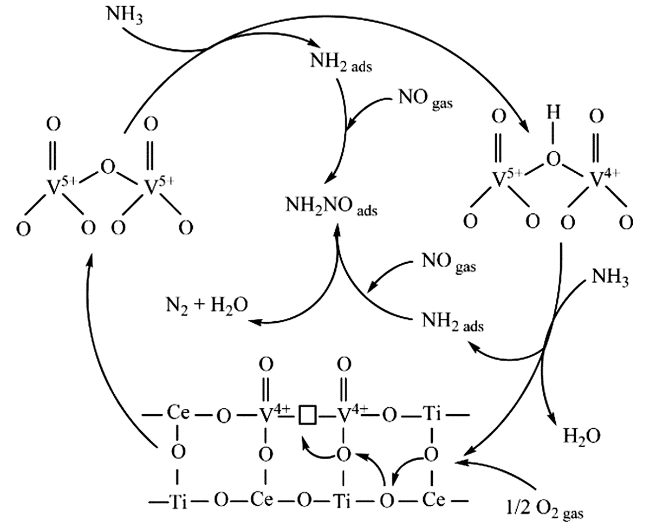
3.5 其他含Ce催化剂
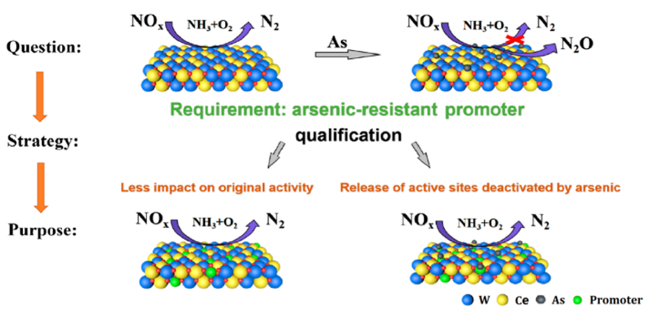
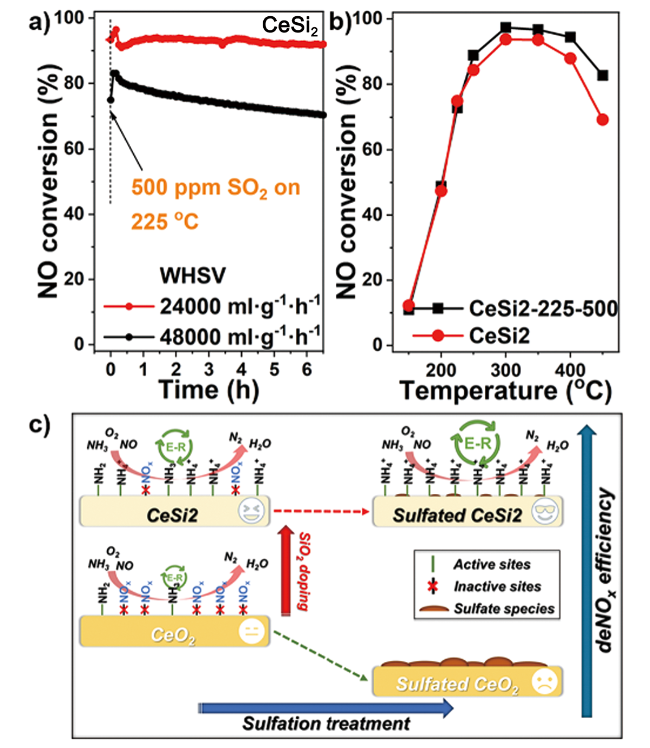
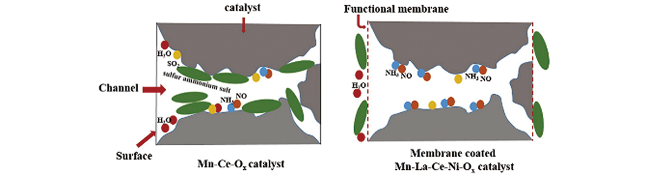
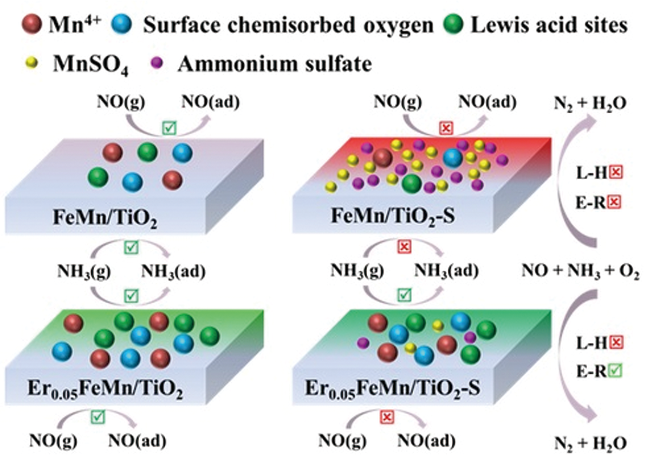
图11 (a)500 ppm SO2对CeSi2性能的影响(反应条件:[NO] = [NH3] = 500 ppm、[O2] = 5 vol%);(b)新CeSi2及在500 ppm SO2条件下使用后的CeSi2性能对比(225 ℃下500 ppm SO2抵抗测试, WHSV = 48 000 mL·g-1·h-1, 使用后的样品命名为CeSi2-225-500);(c)反应机理示意图[82]Fig. 11 (a) NO conversion of CeSi2 in the presence of 500 ppm SO2 at 225 ℃ (reaction condition: [NO] = [NH3] = 500 ppm, [O2] = 5 vol%); (b) NO conversion of fresh CeSi2 and the used CeSi2 (500 ppm of the SO2 resistance test at 225 ℃, WHSV = 48 000 mL·g-1·h-1, the used sample was denoted as CeSi2-225-500); (c) Scheme of the reaction mechanism and reaction performance on fresh and sulfated samples[82]. Copyright 2021, American Chemical Society |
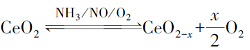
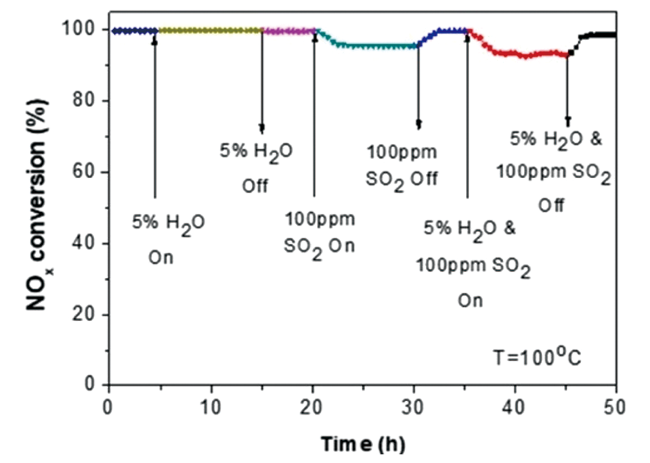
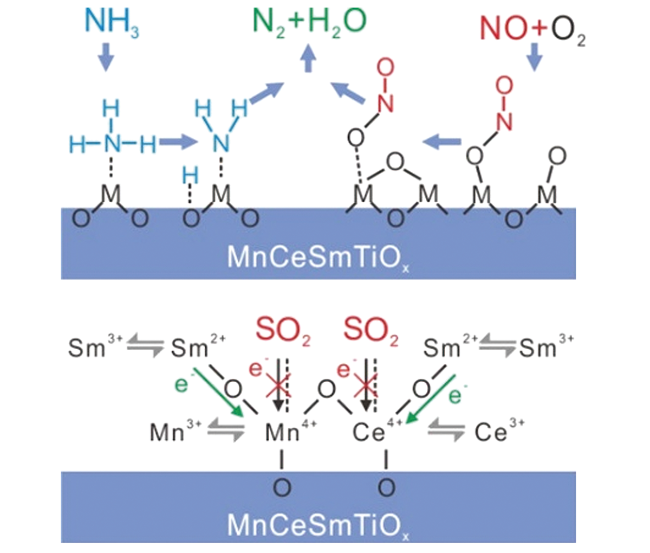

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}