




1 引言
2 有机半导体的物理化学性质
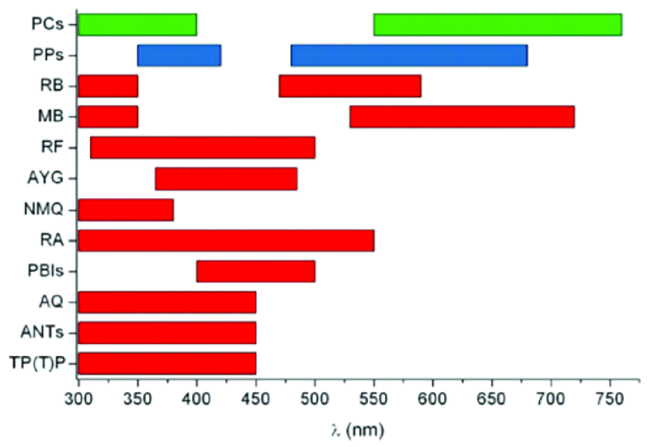
3 光催化过程

4 有机分子自组装

5 有机纳米光催化剂的改性策略
5.1 单一有机纳米光催化剂
5.1.1 零维有机纳米光催化剂
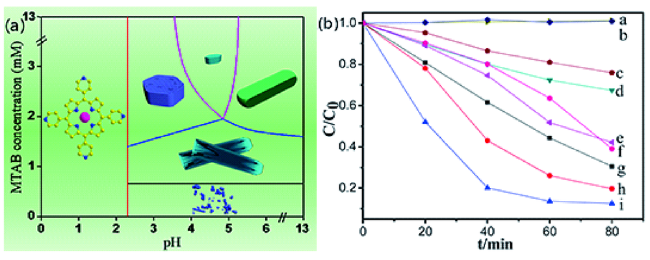
图4 (a)纳米晶形貌随表面活性剂浓度和pH的变化而变化;(b)锌卟啉(ZnTPP)纳米晶的光催化活性。在可见光照射下,四方纳米棒(c)、相同浓度的ZnTPP在DMF中的浓度(d)、相同浓度的ZnTPP在0.01 M的HCl中(e)、直径为80 nm的纳米颗粒(f)、长度为2 μm的六方纳米线(g)、长度为400 nm的六方棒(h)和六方多孔纳米盘(i)用于可见光照射下光降解MO分子。文中还给出了不使用锌卟啉纳米晶(a)和使用商品P25(b)的空白实验结果以供比较[64]Fig. 4 (a) Nanocrystal morphology evolution with the surfactant concentration and pH,(b) Photocatalytic activities of Zn porphyrin(ZnTPP) nanocrystals. Tetragonal nanorods with 200 nm length(c), same concentration ZnTPP in DMF(d), same concentration ZnTPP in 0.01 M HCl(e), nanoparticles with 80 nm diameter(f), hexagonal nanowires with 2 μm length(g), hexagonal rods with 400 nm length(h), and hexagonal porous nanodisks(i) for photo degradation of MO molecules under visible light irradiation. The results from blank experiments, where no Zn Porphyrin nanocrystals were used(a) and commercial P25(b) was used are also presented for comparison[64]. Copyright 2014, American Chemical Society |

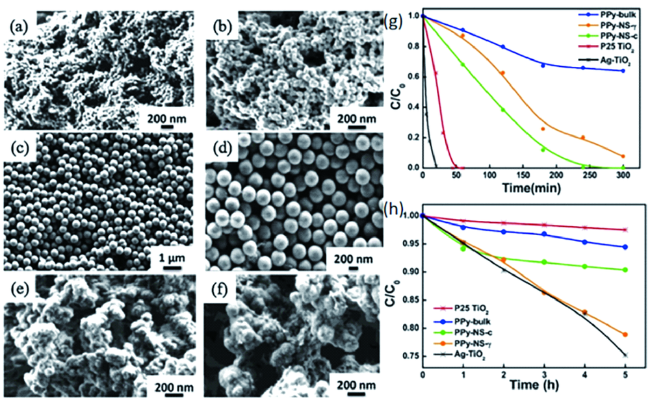
图6 (a,b)PPy-NS-c的SEM图像;(c~d)PPy-NS-γ的SEM图像;(e,f)PPy-bulk的SEM图像;(g)制备的PPy样品在紫外可见光下对苯酚(初始浓度为0.5 mM)的降解率;(h)制备的PPy样品在可见光照射下对苯酚(初始浓度为0.5 mM)的降解率。P25 TiO2在紫外线下显示出高活性,Ag-TiO2在紫外光和可见光下均表现出高活性,在实验中用作对照组衡量PPy样品的催化活性[67]Fig. 6 SEM images of PPy-NS-c(a,b), PPY-NS-γ(c~d), and PPy-bulk(e,f);(g) Degradation rate of phenol(initial concentration: 0.5 mM) under ultraviolet and visible light;(h) Degradation rate of phenol(initial concentration 0.5 mM) under visible light irradiation for the PPy sample prepared. P25 TiO2 showed high activity under ultraviolet light, while Ag-TiO2 showed high activity under ultraviolet and visible light, which was used as the control group to measure the catalytic activity of PPy samples in the experiment[67]. Copyright 2019, Elsevier |
5.1.2 一维有机纳米光催化剂

5.1.3 二维有机纳米光催化剂
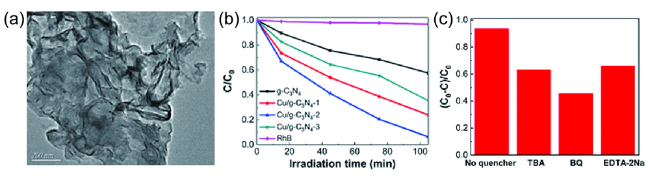
图8 (a)Cu/g-C3N4的TEM图像;(b)纯g-C3N4、Cu/g-C3N4光催化的RhB的降解率;(c)可见光照射下Cu/g-C3N4在有无EDTA-2Na、BQ和TBA时对RhB的光催化活性比较研究[94]Fig. 8 (a) TEM image of Cu/g-C3N4;(b) Photocatalytic degradation of RhB in the presence of pure g-C3N4, Cu/g-C3N4 materials;(c) Photocatalytic activities comparison of Cu/g-C3N4 materials with or without adding EDTA-2Na, BQ and TBA to degrade RhB under visible light irradiation[94]. Copyright 2020, Acta Physico-Chimica. Sinica |
5.2 二元复合有机纳米光催化剂

5.2.1 有机/有机复合纳米有机光催化剂
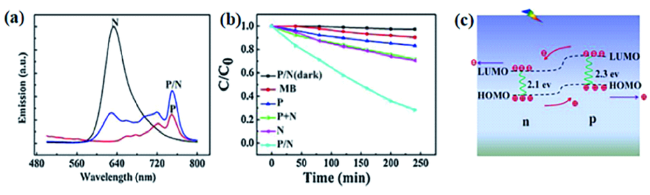
图10 (a)H2TPP(p),CH-PTCDI(n)和H2TPP-CH-PTCDI(p-n)纳米结构的荧光光谱;(b)可见光(λ > 400 nm)下不同样品对亚甲基蓝(MB)的光催化降解;(c)H2TPP-CH-PTCDI纳米复合结构的光催化原理示意图[98]Fig. 10 (a) Fluorescence spectra of H2TPP(p), CH-PTCDI(n) and H2TPP-CH-PTCDI(p-n) nanostructures;(b) photocatalytic degradation of methyl blue(MB) by different samples under visible light(λ > 400 nm) ;(c) Schematic diagram of photocatalysis of H2TPP-CH-PTCDI nanocomposite structure[98]. Copyright 2013, Royal Society of Chemistry |

图11 (a)PTCBI和MPc的分子结构;(b)PTCBI/H2Pc和PTCBI/PbPc的吸收光谱(400~1000 nm);(c) PTCBI/PbPc的长期光电解研究(ITO/PTCBI/PbPc的耐久性能是根据Pt计数器上析出的H2的量来评估的,一个周期的照射时间为1 h)[99]Fig. 11 (a)The molecular structure of PTCBI and MPc;(b)Absorption spectra of PTCBI/H2Pc and PTCBI/PbPc (400~1000 nm);(c)A long-term optoelectronic solution study of PTCBI/PbPc(the durability of ITO/PTCBI/PbPc was assessed by the amount of H2 precipitated on the Pt counter and the exposure time for one cycle was 1 hour)[99]. Copyright 2017, Elsevier |
5.2.2 有机/炭复合纳米有机光催化剂
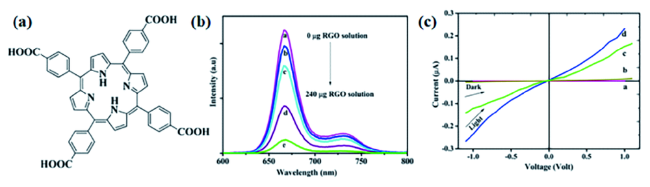
图13 (a)5,10,15,20-四(4-羧基苯基)卟啉(TCPP)的化学结构;(b)不同浓度的rGO(a-0 μg,b-60 μg,c-100 μg,d-150 μg和e-240 μg)时,TCPP NR-rGO复合材料的光致发光光谱;(c)TCPP NR的暗电流a和光电流c;TCPP NR-rGO复合系统的暗电流b和光电流d[105]Fig. 13 (a) The chemical structure of 5,10,15,20-four(4-carboxyphenyl) porphyrin(TCPP) ;(b) Photoluminescence spectra of TCPP NR-rGO composites at different concentrations(a-0 μg, b-60 μg, c-100 μg, d-150 μg and e-240 μg) ;(c) Dark current a and photocurrent c of TCPP NR; Dark current b and photocurrent d of TCPP NR-rGO composite system[105]. Copyright 2016, American Chemical Society |

图14 (a)使用NaBH4还原制备rGO/pCN的静电自组装流程图;(b)rGO、纯g-C3N4、pCN、15rGO/pCN和15rGO/CN光照10 h后CH4的总产量[107]Fig. 14 (a)Schematic mechanism for fabricating rGO/pCN heterojunction via an electrostatic self-assembly strategy followed by reduction with NaBH4;(b)Total yield for CH4 production over rGO, pure g-C3N4, pCN, 15rGO/pCN, and 15rGO/CN photocatalysts after 10 h of illumination[107]. Copyright 2015, Elsevier |
5.2.3 有机/金属复合纳米有机光催化剂
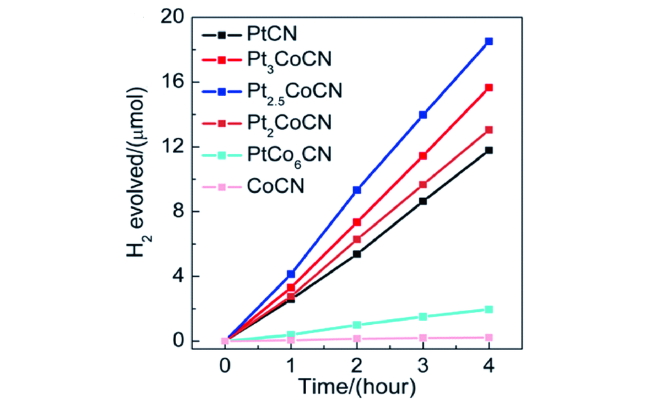
5.2.4 有机/无机复合纳米有机光催化剂
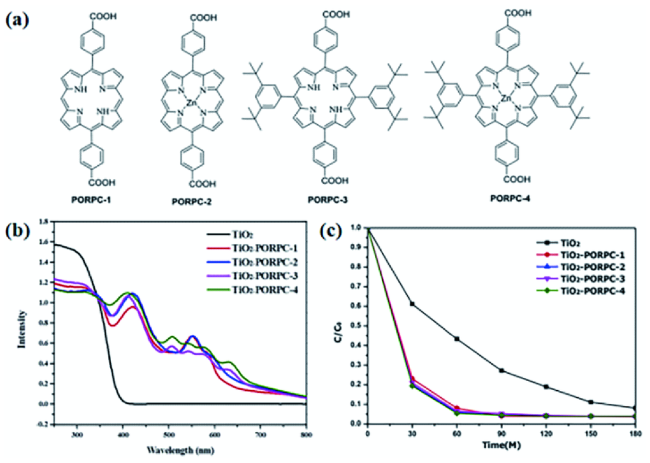
图16 (a)四种卟啉分子的结构;(b)纯TiO2和四种TiO2-卟啉复合光催化剂的可见光吸收光谱;(c)纯TiO2和四种TiO2-卟啉复合光催化剂在可见光照射下MB的光催化降解曲线[115]Fig. 16 (a)The structure of the four porphyrin molecules;(b)Visible absorption spectra of pure TiO2 and four kinds of TiO2-porphyrin composite photocatalysts;(c)Photocatalytic degradation curves of MB with pure TiO2 and four kinds of TiO2-porphyrin composite photocatalysts under visible light irradiation[115]. Copyright 2019, Elsevier |
5.3 多元复合有机纳米光催化剂
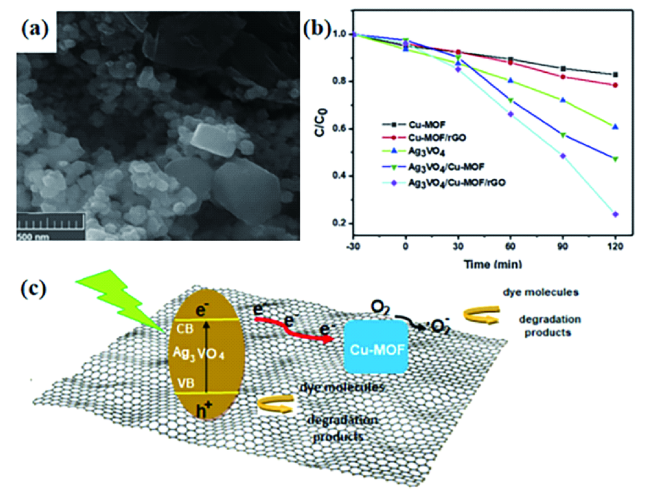
图17 (a)Ag3VO4/Cu-MOF/rGO的FESEM图片;(b)在不同催化剂存在下AB92光降解率C/C0图像;(c)Ag3VO4/Cu-MOF/rGO在可见光照射下的光催化机理和电荷转移方案[126]Fig. 17 (a) FESEM image of Ag3VO4/Cu-Mof/rGO;(b) Image of AB92 photodegradation rate C/C0 in the presence of different catalysts;(c) Photocatalytic mechanism and charge transfer scheme of Ag3VO4/Cu-Mof/rGO under visible light irradiation[126]. Copyright 2020, Elsevier |
6 有机纳米光催化剂的应用
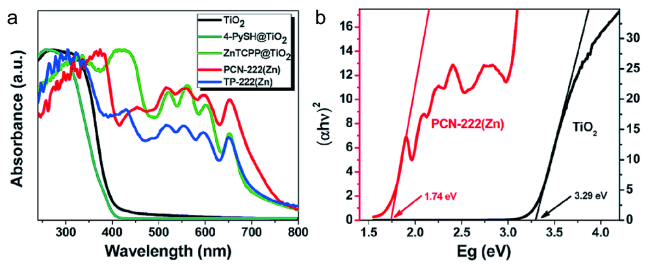
图18 (a)TiO2、4-PySH@TiO2、ZnTCPP@TiO2、PCN-222(Zn)和TP222(Zn)的紫外-可见漫反射光;(b)根据(αhv)2-Eg线计算了TiO2和PCN-222(Zn)的带隙[138]Fig. 18 (a) UV-visible diffusive reflectance spectra of the as-prepared samples: TiO2, 4-PySH@TiO2, ZnTCPP@TiO2, PCN-222(Zn) and TP-222(Zn);(b) Calculation of the band gaps of TiO2 and PCN-222(Zn) based on the(αhv)2-Eg curves[138]. Copyright 2017, Royal Society of Chemistry |

图19 (a)g-C3N4-NiFeP的TEM图像;(b)不同样品的平均析氢率;(c)TRPL谱;(d)EY敏化的g-C3N4,CN/FEP/g-C3N4和CN/FeNiP/g-C3N4复合材料的光电流响应[143]Fig. 19 (a) TEM image of g-C3N4-NiFeP;(b) average H2 evolution rate over different samples;(c) TRPL spectra and(d) photocurrent responses of EY-sensitized g-C3N4, CN/FeP/g-C3N4, and CN/FeNiP/g-C3N4 composites[143]. Copyright 2019, Elsevier |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
7 结论及展望
表1 纳米有机光催化剂的制备、形貌及应用[24⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓~35,53⇓⇓ ~56,129⇓⇓⇓⇓⇓⇓ ~136]Table 1 Preparation, morphology and application of nano organic photocatalyst[24⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓~35,53⇓⇓ ~56,129⇓⇓⇓⇓⇓⇓ ~136] |
| Photocatalyst | Preparation method | Structure and morphology* | Application and efficiency | ref |
|---|---|---|---|---|
| H-type aggregated perylenete- tracarboxylic diimide(H-PDI); J-type aggregated perylenete tracarboxylic diimide(J-PDI) | pH hydrogelation method | Nanofibers(SSA: 10.05 m2·g-1) Nanorods( SSA: 16.02 m2·g-1) | phenol photodegradation(50%, 4 h) phenol photodegradation(negligible) | 54 |
| perylene diimide/nanosilica(SN-PDI) | covalently anchoring | Nanospheres( SSA: 39.9 m2·g-1) | Decabromodiphenyl ether(BDE209) photodegradation(100%, 0.5 h) | 55 |
| perylene imide/Bi2WO6 (PDI/Bi2WO6) | dual transfer approach | Nanosheets( SSA: 16.633 m2·g-1) | BPA photodegradation(99.7%, 3 h) | 56 |
| 20-tetrakis[p-(3-N-triethoxysilylpropylureido)phenyl]porphyrin(TiO2/POR-Si) | Sol-gel cogelation | Crystallite( SSA: 180 m2·g-1) | P-nitrophenol(PNP) photodegradation(65%, 24 h) | 31 |
| Tetrahydroxyphenyl zinc porphyrin-TiO2(ZnTHPP-TiO2); Tetrahydroxyphenyl zinc porphyrin/TiO2(ZnTHPP/TiO2) | Solvothermal in situ method; Impregnation method | Nanospheres(size: 30~40 nm) | MB photodegradation(75.6%, 100 min); MB photodegradation(61.2%, 100 min) | 24 |
| Iron(Ⅲ) meso-tetra(4-carboxyphenyl) porphyrin/TiO2 nanotubes(FeTCPP-TNTs) | hydrothermal and heating reflux process | Nanospheres (size: 18.26 nm, SSA: 309.45 m2·g-1) | MB photodegradation(90%, 2 h) | 35 |
| Tetrakis(4-carboxyphenyl)porphyrin /ZnFe2O4@polythiophene(TCPP/ZnFe@PTh) | photosensitize | Clusters | MO photodegradation(94%, 3 h) | 34 |
| 6- 5- dicarbaldehyde(CuTAPP-CMP-OH) | Microwave-assisted | Nanospheres(SSA 223.6 m2·g-1) | RhB photodegradation(98%, 3 h) | 26 |
| polyphenylporphyrin benzobisoxazole P(PPor-BBO) | polycondensation | Nanospheres(size: 90 nm) | RhB photodegradation(98%, 2.5 h) | 30 |
| 20-meso-tetra-(para-amino)-phenyl- porphyrin(FST-g-TAPP) | Graft modification | Clusters(75.127 m2·g-1) | RhB photodegradation(96.26%, 1 h) | 27 |
| Benzobisoxazole-linked porphyrin-based fully conjugated microporous polymers based on metalloporphyrin(BBO-NiPor-CMP) | polycondensation reaction | Sheet clusters(SSA: 238 m2·g-1) | RhB photodegradation(100%, 2.5 h) | 33 |
| Carbon nitride/cobalt tetra-phenyl-porphyrin(CN/Co(Ⅲ)TPP) | Self-assembled | Lamellar(pore volume: 0.254 cm3·g-1) | Nicotinamide cofactors(NADH) regeneration(87.9%, 1 h) | 25 |
| cobalt metallated aminopor-phyrin(GO-Co-ATPP) | Solvothermal method | Nanospheres(SSA: 13.971 m2·g-1) | Formic acid conversion(96.49 μmol, 2 h) | 28 |
| g-C3N4 loaded with carbon dots/tetra (4-carboxyphenyl)porphyrin iron(Ⅲ) (g-C3N4-Cx/FeTCPP) | mechanical mixing | Nanosheets(SSA: 77.5~85.5 m2·g-1) | CO evolution(28.3 mmol g-1, 6 h) H2 evolution(71.1 mmol g-1, 6 h) | 32 |
| Zinc-porphyrin/platinum deposited hi- erarchical porous TiO2(LG5/PHPT) | D-π-A approach | Clusters | H2 evolution(4196 μmol·g-1·h-1) | 29 |
| 4-pyrrolopyrrole dione(DPP-CN); 4-pyrrolopyrrole dione(DPP-CI); 4-pyrrolopyrrole dione (DPP-PN) | Condensation reaction | Strip(size: 176.8 nm) Flake(size: 316.9 nm) Strip(size: 285.8 nm ) | H2 evolution(569.26 μmol·g-1·h-1) H2 evolution(361.84 μmol·g-1·h-1) H2 evolution(Almost none) | 53 |
| Pt@CeO2/three-dimensional porous g-C3N4(Pt@CeO2/3DCN) | Calcination method | Cubes/sheets(SSA: 61.67 m2·g-1) | CO evolution(4.69 μmol·g-1·h-1) CH4 evolution(3.03 μmol·g-1·h-1) | 129 |
| 4- benzenedicarboxylate)/TiO2(CPO-27-Mg/TiO2) | Hydrothermal Self-assembly method | Spindle/nanospheres(size: 300~500 nm) | CO evolution(40.9 μmol·g-1) CH4 evolution(23.5 μmol·g-1) | 130 |
| Cs2AgBiBr6@g-C3N4 (CABB@g-C3N4) | In situ assembly strategy | Thin shell(size 2~3 nm) | CO2 reduction(2.0 μmol·g-1·h-1) | 131 |
| Zr based MOFs/carbon nanotubes(UIO-66-NH2/CNTs) | Hydrothermal method | spheres/rods(SSA: 642.5 m2·g-1) | CO2 reduction(28.8 μmol, 4 h) | 132 |
| 4'-(PO3H2)2-bipy)]Br2-TiO2-CO2-reducing enzyme(RUP-TiO2-CODH) | modified with a photosensitizer | Nanospheres(size: 21 nm) | CO evolution(5 μmol, 4 h) | 133 |
| MOF-525(Zr6O4(OH)4(TCPP-H2)3)-Co(MOF-525-Co) | incorporation of coordinatively unsaturated single atoms | Nanosheets | CO evolution(200.6 μmol·g-1·h-1) CH4 evolution(36.76 μmol·g-1·h-1) | 134 |
| MIL-101=Fe-containing MOFs (NH2-MIL-101) | amine-functionalized | Clusters | CO2 adsorption(34.0 cm3·g-1) | 135 |
| Au@PtAg/2-Methylimidazole zinc salt(Au@PtAg@ZIF-8) | encapsulation approach | Core/hell(SSA: 1325 m2·g-1) | CO evolution(14.5 μmol·g-1·h-1) | 136 |
*SSA means specific surface area |