




1 引言
2019 年发布的《 BP 世界能源统计年鉴》显示,随着全球能源需求量的加速增长,2018 年全球碳排放量增加了 2%,高于历史平均水平的 1.4%[1]。碳排放量的增长,以及酸雨、雾霾等环境问题的日益严重,促进了清洁能源的开发和利用。燃料电池(Fuel cells, FCs)具有反应产物简单,绿色、安全的特点。在各种燃料电池中,质子交换膜燃料电池(Proton exchange membrane fuel cells, PEMFCs)具有能量转化率高、功率密度高、冷启动快等优点,被视为最具有商业价值的燃料电池,从而获得了广泛的关注[2]。根据工作温度的不同,质子交换膜燃料电池可分为传统的低温质子交换膜燃料电池(LT-PEMFCs)和高温质子交换膜燃料电池(HT-PEMFCs),前者的工作温度一般在 80 ℃ 以下,而后者的工作温度则在 100~200 ℃。与低温相比,PEMFCs 在高温下运行具有提高催化剂对 CO 的耐受性,加速电极反应动力学,简化水、热管理,降低成本等优势[3⇓~5]。
质子交换膜(Proton exchange membranes, PEMs)是 PEMFCs 的核心部件,为保证电池的高效运行,PEMs 需要具有高质子传导率、低燃料透过率、良好的绝缘性,及良好的化学稳定性、机械稳定性、结构和形态稳定性。
为了制备适用于 HT-PEMFCs 的质子交换膜,人们对全氟磺酸类 PEMs 进行改性,例如引入亲水性无机金属氧化物来解决高温下水流失的问题[8],或引入杂多酸来提高高温下的质子传导能力[9],但质子交换膜的工作温度仍无法提高到 120 ℃ 以上。其他基于磺酸的 PEMs 如磺化聚芳醚酮、磺化聚芳醚砜、磺化聚酰亚胺、磺化聚苯醚等材料也只适用于 LT-PEMFCs。本质上的原因是磺酸基在失水状态下无法电离,缺少质子导体,即便是被视作发展前景最好的聚苯并咪唑(Polybenzimidazole, PBI)材料,掺杂磺酸后同样需要相对湿度高于 50% 才能获得比较高的质子电导率[9],不能满足高温无水的需要。
目前关于磷酸掺杂的高温质子交换膜的研究已经取得了丰富的成果,为提高膜的综合性能,涌现了大量针对基材高分子以及膜本身的改性研究。此外,基材高分子的选择范围正不断拓展,MOFs、COFs 等作为晶态质子导体也在本领域崭露头角。因此本文结合 PEMs 中质子传输机制以及磷酸基高温 PEMs 目前存在的问题及挑战, 对近年来包括 PBI 类、非 PBI 类在内的磷酸掺杂高温质子交换膜的改性研究进行了较全面的综述,并由多孔 PBI 引入,梳理总结了 MOFs、COFs 等新型多孔材料在 PEMs 领域的应用,这一类材料种类繁多,可设计性强,虽然目前相关研究仍处在初始阶段,但已显示出用于 HT-PEMFCs 的潜力。最后,简要总结并评价了各种改性手段的优劣,指出了MOFs、COFs 在本领域的应用前景,旨在对未来的研究有所帮助。
2 质子交换膜的质子传导机理
质子传导的机理一般分为两种,即运载机理(Vehicular mechanism)和跳跃机理(Hopping mechanism/Grotthuss mechanism)[15]。
运载机理认为,质子与质子载体(一般为水分子)结合,并通过膜的微观结构,从阳极向阴极定向移动来完成质子传导[17]。跳跃机理认为质子的传递是基于膜内的氢键网络,质子从某一载体上通过氢键的伸缩振动跳跃到相邻的质子载体,原来的质子载体重新恢复接受质子的能力,从而形成质子的定向连续传递[18]。跳跃机理和运载机理能同时存在并相互影响,例如 Nafion 膜中就同时存在着两种机理[19]。一般情况下,当膜内具有充足的水分和氢键位点时,易形成连续的氢键网络,主要以 Grotthuss 机理传递质子,传递效率较高。相反则主要遵循运载机理,需要克服更高的能垒,质子传导效率较低[20]。Grotthuss 机理质子传导的活化能为 14~40 kJ/mo1,可作为质子传导机制的判据[21,22]。
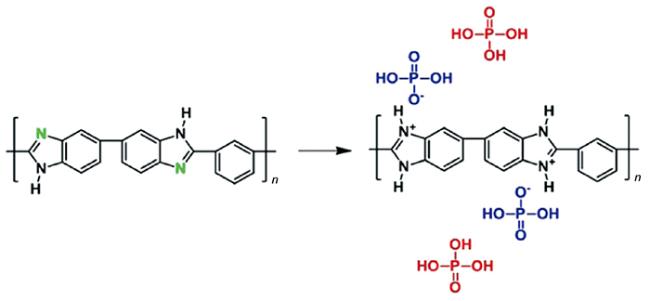
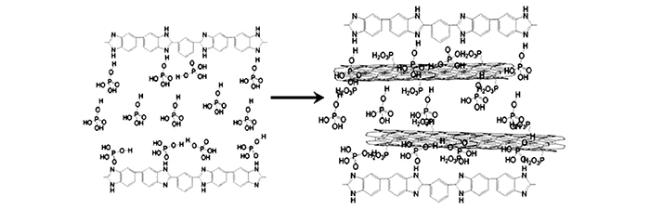
 ),可以与磷酸分子发生酸碱相互作用,从而产生“键合酸”,同时磷酸分子也可以通过氢键相互作用与聚合物骨架和其他磷酸分子连接,实际工作中磷酸掺杂量往往很高,这时分子链间存在大量的“游离酸”,与聚合物骨架共同形成氢键网络[
),可以与磷酸分子发生酸碱相互作用,从而产生“键合酸”,同时磷酸分子也可以通过氢键相互作用与聚合物骨架和其他磷酸分子连接,实际工作中磷酸掺杂量往往很高,这时分子链间存在大量的“游离酸”,与聚合物骨架共同形成氢键网络[文献中将每个聚苯并咪唑重复单元吸附的磷酸分子总数目称为磷酸掺杂水平(Acid doping level, ADL),如图 1 中的 ADL 为 4,蓝色表示键合酸,红色表示游离酸。
3 磷酸基高温质子交换膜面临的问题及挑战
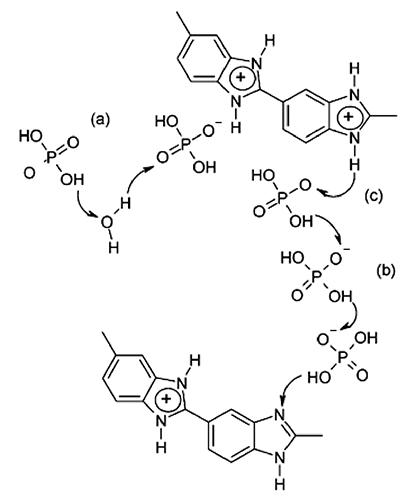
磷酸掺杂的高温质子交换膜,实质上是一种酸碱复合膜,由含有碱性基团(如亚胺、酰胺、咪唑基团等)的聚合物作为基材,提供磷酸吸附位点,并赋予材料加工性。磷酸分子是质子载体,其含量直接决定质子传导性能。同时磷酸相当于增塑剂,吸附适量的磷酸,能够增大高分子链间的距离,减弱分子间作用力,从而增进膜的柔韧性,改善和提高膜的脆性和加工性能。但是,高的吸酸率也意味着大的尺寸溶胀和机械强度的降低,过高的磷酸含量甚至会导致膜的变形和破碎,这是磷酸膜研究中的一个重点和难点。
另一个重要问题是磷酸的流失。Lee等通过实验研究,认为磷酸的流失主要是阴极侧存在的水蒸气与液态水引起的[28],PEMFCs运行过程中,大量水在阴极产生并附着,一定程度上会吸收膜中的磷酸分子,在电池长期高温运行状态下,游离的磷酸不断向外扩散而流失,不仅降低了质子传导能力,还会腐蚀电池的其他部件。
4 磷酸基高温质子交换膜的改性
为了制备质子传导能力与机械性能兼顾的磷酸基质子交换膜,研究者们对基材高分子进行了多种改性。由于目前 PBI 类质子交换膜仍被视作最有潜力的 HT-PEMFCs 候选膜材料,且相关研究最为深入和广泛,因此接下来将分为 PBI、非 PBI 两部分,前一部分对磷酸基高温质子交换膜的改性手段进行分类总结,后一部分补充介绍其他类型的高温质子交换膜。
4.1 PBI 类高温质子交换膜
自1961年聚苯并咪唑类聚合物首次被合成以来,聚[5'-二苯并咪唑](通常称为m-PBI)得到了最广泛的研究与应用,也是商业化最早、最成功的 PBI,其具有非常优异的耐热性、防火耐燃性、高的机械性能,以及极好的耐化学药品、耐苛刻环境性能,在工业、军需、民用等各方面得到广泛应用[29]。
4.1.1 主链改性的聚苯并咪唑质子交换膜材料
PBI主链的改性是从分子结构设计出发,将某些特定基团引入到聚合物主链。在实际的工作中,常同时引入多种基团,以下分为两部分进行介绍。
(1)引入柔性基团、大体积侧基和非共面结构
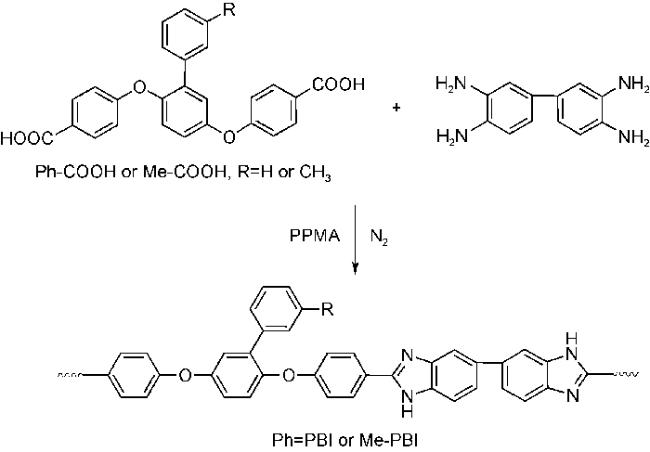
将柔性基团如醚键(—O—)、羰基(—CO—)、砜基(—SO2—)、六氟代异丙基(—C(CF3)2)[34⇓~36],大的侧基[37] 或非共面结构[38,39]引入聚合物,能降低高分子链的刚性和对称性,防止高分子链的紧密堆积,提高溶解性。Liu等[40]合成了含非对称侧基和醚键的聚苯并咪唑Ph-PBI 和 Me-PBI(如图3所示),尽管分子量分别高达 104 kDa 和 118 kDa,却可以在室温下以 10% 的浓度完全溶解于DMSO、DMF、DMAc、NMP等有机溶剂中。磷酸掺杂后,膜在200 ℃下的质子传导率分别达到 0.217和 0.165 S·cm-1,拉伸强度分别为5.5和6.1 MPa。
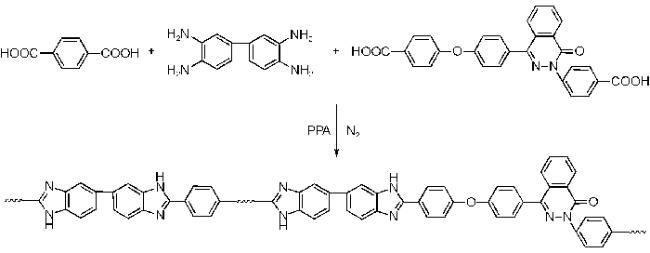
(2)引入其他功能基团
在 PBI 中引入磺酸基团,可以结合高温膜和低温膜共同的优势。根据报道,磷酸掺杂的磺化聚苯并咪唑(SPBIs)比相应的非磺化聚苯并咪唑具有更高的质子传导率[43,47]。磺化聚苯并咪唑膜的制备方法主要可以分为用磺化单体合成聚合物(先磺化)和将聚合物的膜磺化(后磺化)两种。先磺化可以设计出目标结构的聚合物,且可以精确控制磺化度,但是多数磺化单体都不能商业购买,后磺化非常简单,但聚合物的反应活性都很低,例如 m-PBI 及聚(2,5-苯并咪唑)(ABPBI)都需要高的反应温度(450~475 ℃)来磺化,从而会得到交联以及低磺化度的脆性膜[48]。聚[5'-联苯并咪唑](OPBI)由于醚键的给电子效应,反应活性高一些,可以在 80 ℃ 下采用浓硫酸或发烟硫酸进行磺化。Fang等[48]将OPBI后磺化,制备了磺化聚苯并咪唑SOPBI,其在DMSO中有良好的溶解性,具有优异的热性能、良好的成膜性和机械性能,但是他们并没有研究磷酸的掺杂行为以及磷酸掺杂的SOPBI的质子传导率。
Quartarone等[44]使用先磺化的方法制备了一系列具有不同磺化度的 SPBIs 膜(如图5所示)。相同实验条件下,磺化程度低的 SPBIs 膜具有更高的 ADL 和质子传导率,Zhang等[49]的研究工作中也发现了相同的情况,这是由于咪唑环的碱性位点与磺酸基发生了分子内相互作用,不能再为磷酸的吸附和解离做出贡献。然而,在相同的 ADL 下,磺化程度高的 SPBIs 膜具有更高的质子传导率,且所有的SPBIs都比未磺化的PBI膜质子传导率高出很多,这说明磺酸基对质子的传导做出了贡献[50]。SPBIs 膜组装的H2/空气燃料电池在 150 ℃ 的最大功率密度约为 320 mW·cm-2。
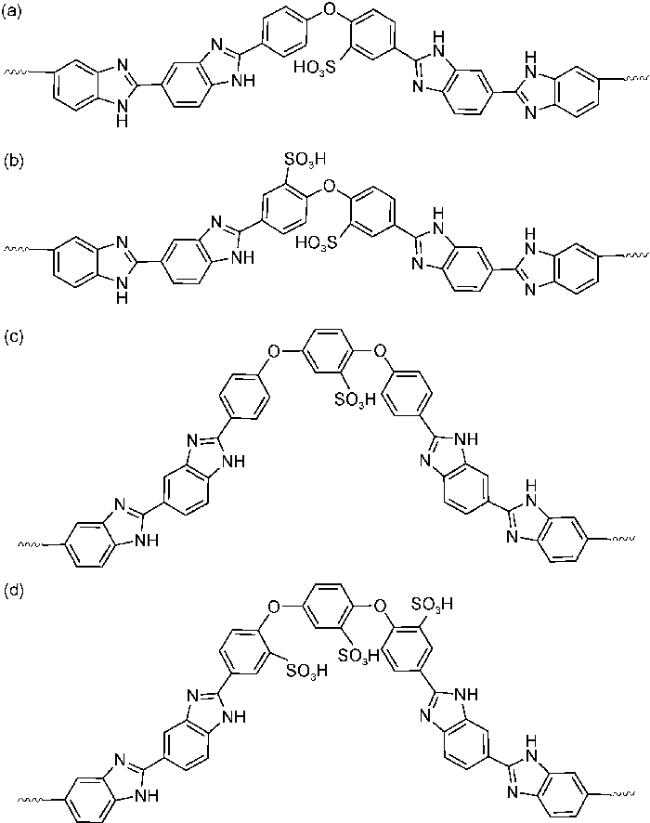
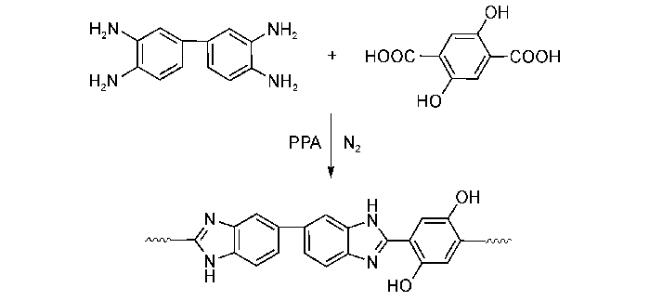
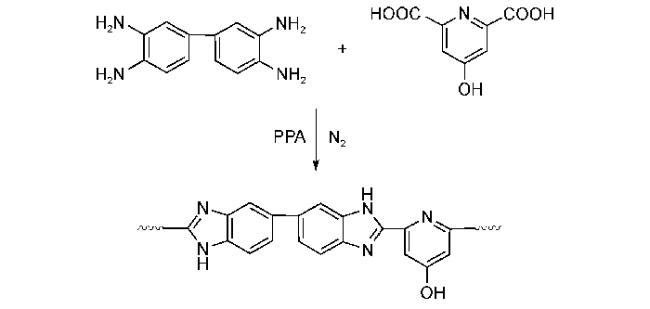
4.1.2 掺杂型聚苯并咪唑质子交换膜材料
通过向高分子中掺杂无机物、质子导体等添加物,可制备掺杂型复合膜,进而提高膜的热稳定性、电导率和机械性能等。通常,无机相的引入能减小磷酸对膜的塑化作用,进而增加膜的机械强度,但对于有机-无机复合膜来说,必须考虑无机相在聚合物膜中的分散性和相容性,且高温下无机纳米颗粒还可能会发生聚集。基于以上问题,无机填料通常被设计成纳米或微米尺度,且表面引入各种有机基团进行化学修饰。有机小分子作为 PBI 的掺杂物相容性更好,但由于稳定性较差而实例较少。按照掺杂物的类型,可分为以下四类[51]:
(1)掺杂惰性吸湿氧化物
Han 等[53]对上述工作进行改进。通过表面修饰方法,先后在 TiO2纳米颗粒表面引入苯基和磺酸基,合成了磺化的二氧化钛纳米颗粒(s-TiO2),并用来修饰 m-PBI。同样地,2 wt% s-TiO2 的复合膜获得最高的磷酸掺杂水平(ADL=12.1),150 ℃ 的质子传导率为 0.096 S·cm-1,峰值功率密度达 621 mW/cm2,比纯 PBI 膜高出 30%。对比可知 s-TiO2的改性效果比 TiO2 更好。
(2)掺杂固体酸/盐
杂多酸(HPA)是具有高传导性和酸性的固体酸,质子传导主要遵循 Grotthuss 机理[60]。Scott 等[57]制备了磷酸掺杂的 PWA/PBI 和 SiWA/PBI 复合膜,含 40% SiWA 的复合膜在 150 ℃ 的质子传导率达到 0.1774 S·cm-1。他们又合成了四种杂多酸铯盐纳米粒子[59]:CsPOMo、CsPOW、CsSiOMo 和 CsSiOW,并与 PBI 复合制备质子交换膜。磷酸掺杂的复合膜均比纯 PA/PBI 膜具有更高的质子传导率和电池性能,且质子传导率随着复合膜中 CsHPA 含量的增加而提高。30 wt%CsPOMo/PBI 薄膜在 150 ℃ 无水条件下表现出 0.12 S·cm-1的最大传导率,同温度下 H2/O2 燃料电池的最大功率密度约为 0.6 W·cm-2,但机械强度稍低。
磷酸氢锆(ZrP)也能提高复合膜的质子传导性能,Xie 等[58]将 ZrP 与 SPBIs 复合,SPBI-10-Z3 复合膜 180 ℃ 的质子传导率为 0.08 S·cm-1,比原始的 PA/ SPBIs 膜提高了 20%。
(3)掺杂碳基材料
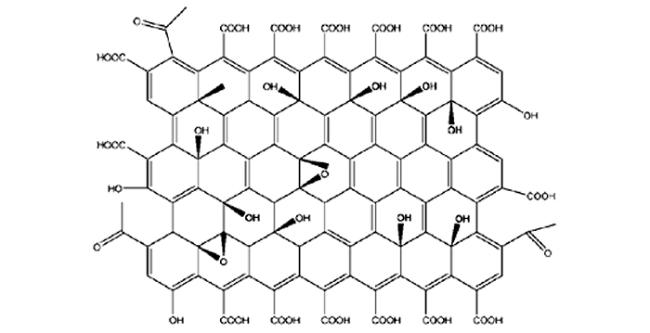
He 等[65]将三唑接枝的氧化石墨烯 MGO 应用于 PBI,不仅提高了膜的拉伸强度,还可以促进质子的传输。磷酸掺杂的PBI/MGO膜在180 ℃ 无水条件下表现出 0.135 S·cm-1的质子传导率。
(4)掺杂有机物
4.1.3 交联或接枝型聚苯并咪唑质子交换膜材料
交联网络的存在会限制聚合物分子间的体积,同时也限制聚合物分子的移动,降低聚合物膜的尺寸变化,合理的结构设计能够在不降低质子传导率的情况下,提高膜的尺寸稳定性和机械稳定性。根据分子间相互作用的类型不同,可将常见的交联类型分为共价交联和离子交联。
(1)共价交联
共价交联即是在高聚物链间形成共价化学键。有些文献将咪唑 NH 与亲核基团(如卤化物、环氧化物)发生 SN2 反应的交联称作共价交联[69],文献中这样的交联占大多数。此外还存在其他的交联剂和交联方法,可以不以 NH 为交联点。
He 等[69]使用小分子 1,3,5-三溴甲基苯(B3Br)与 1,3,5-三(溴甲基)-2,4,6-三乙基苯(Be3Br)作交联剂,制备了两种 PBI 交联膜。其中 Be3Br-7.5%/11.6PA 膜在 180 ℃ 未加湿条件下质子传导率达 0.095 S·cm-1,室温拉伸强度为 8.5 MPa。总的来讲,交联结构稍稍提高了 m-PBI 的磷酸掺杂率与质子传导率,而在机械强度上提高比较明显。
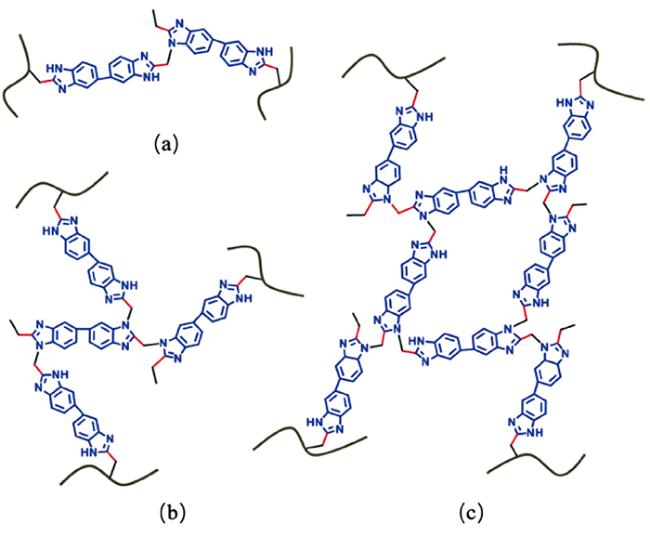

机械强度与质子传导率得以同步提高。磷酸掺杂的 QOPBI-15 膜(交联剂含量 15 wt%)在 160 ℃ 无水条件下的质子传导率为 0.049 S·cm-1,最为优异的是机械性能,拉伸应力高达 27 MPa。
Wang等[73]合成了支化型 F6PBI,接下来用聚-双酚 A 型苯并口恶嗪 p(BA-a) 作为交联剂,得到交联型聚苯并咪唑。交联剂 p(BA-a) 本身含有 N 原子,且长链结构可增大自由体积,因此随着交联剂含量增加,PA 的吸附量也增加。提高质子传导率的同时,膜的机械性能也得到提升,支化交联膜 C15F6-R2-6 在 160 ℃ 无水条件下的质子传导率为 0.073 S·cm-1,拉伸应力为 17 MPa,用 C10F6-R2-6 组装氢氧燃料电池,160 ℃ 的峰值功率密度达 690 mW·cm-2。
此交联不以 NH 为交联点,而是以 BA-a 分子 N 和 O 之间的亚甲基进攻 F6PBI 的富电子位点,发生亲电取代。交联位点在图 12 中标出。
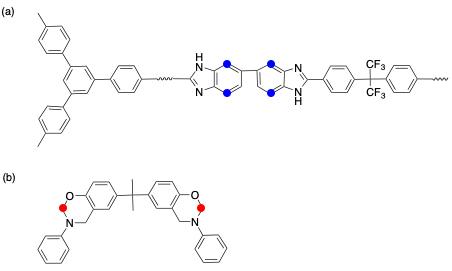
(2)离子交联
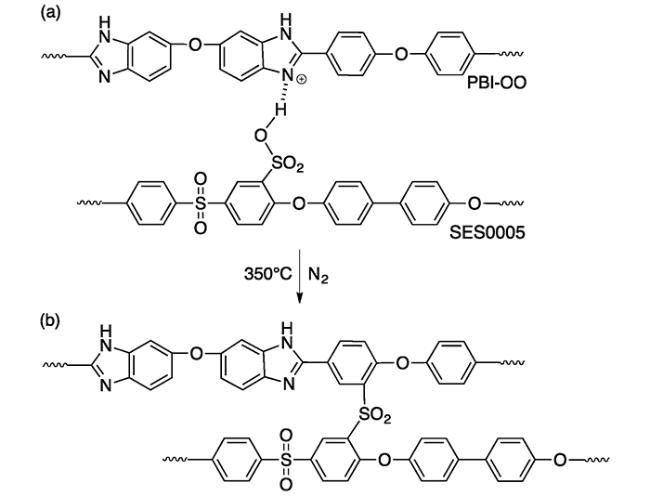
热固化后,膜的磷酸吸附量有所降低,但质子传导率与组装电池的性能却提高,尤其是电池运行的耐久性提高很大,膜的拉伸强度变化不大,均在 10 MPa 以上。共价交联膜 c-BM 1(5% SES0005, 95% PBI-OO)在 160 ℃ 的质子传导率为 0.260 S·cm-1,比同类型不同比例的共价交联膜高出一倍,原因未明,他们猜测可能是低浓度的酸性聚合物导致了利于磷酸吸附的微观结构的形成。
值得注意的是,PBI-OO 的醚结构使得苯环更加富电子,有利于共价交联的进行,而 PBI-OO 自身的稳定性就较差,甚至在 150 ℃ 的 PA 中会完全溶解。如果可以将 m-PBI 与 SES0005 共混后热固化,膜的稳定性将会更好,但此做法的可行性还没有得到验证。
(3)接枝改性
在聚合物主链上通过化学反应引入含碱性基团(如季铵盐、咪唑、吡啶、吡咯烷酮等)的侧链,不仅提供了新的碱性位点,还破坏了分子链的紧密堆积,增大了自由体积,因此可以直接提高磷酸掺杂水平。例如 Liu 等[76]利用 N-取代反应将苯并咪唑接枝到芳醚型聚苯并咪唑上,在 200 ℃ 未加湿的条件下,最高质子传导率达 0.212 S·cm-1,160 ℃ 下氢氧燃料电池的能量密度峰值为 443 mW·cm-2,综合性能较好,但与接枝前相比,机械强度有所下降。
为了解决磷酸流失的问题,一种策略是通过化学方法将磷酸分子固定在聚合物分子链上,例如通过接枝固定。Yu 等[47]合成了乙烯磷酸接枝的聚苯并咪唑 PBI-EPA。接枝膜自身的机械强度非常差,在 70 ℃ 的浓磷酸中就会溶解,因此与 PBI 混合制备质子交换膜。PBI/PBIEPA 质量比 8∶2 的共混膜 PA 掺杂量最高,达 350 wt%,质子传导率为 0.068 S·cm-1。这种接枝方法虽然固定了磷酸分子,但是牺牲咪唑氢,本质上不利于质子的传递。此外还有将聚乙烯磷酸(PVPA)接枝到 PBI 主链的研究[77],接枝膜的最高质子传导率在 120 ℃ 和 100% RH 达到 0.035 S·cm-1,湿度降到 20% 时仅 0.008 S·cm-1,不适合用作高温质子交换膜。
4.1.4 共混型聚苯并咪唑质子交换膜材料
(1)PBI 与磺化聚合物共混
磺化聚合物通常在有机溶剂中具有良好的溶解性,且机械性能好,价格低廉,可以弥补 PBI 在溶解性能上的不足。磺化聚芳醚类聚合物与 PBI 类聚合物可发生离子交联作用,对提高质子传导率有一定帮助。Bjerrum 等[78]制备了磺化聚砜 SPSF 与 PBI 的共混膜。PBI 质量分数为 75% 或 20% 的混合膜都具有比纯 PBI 膜更高的质子传导率,而前者更高。控制变量的情况下,SPSF 的磺化度越高,质子传导率越高,证明磺酸基为质子传导做出了贡献。当 SPSF 的相对含量较低或 SPSF 的磺化度较低时,PBI 和 SPSF 的相容性变差,出现分相的情况,这也是共混膜需要特别注意的问题。
Zheng 等[79]用 16% 和 32% 两种磺化度的 SPEEK 合成了 SPEEK/ABPBI(质量比1∶5)共混膜。磷酸掺杂后,(SPEEK16%)/ABPBI 膜在100 ℃下质子传导率为 0.109 S·cm-1,是单纯 ABPBI 膜在该条件下质子传导率的 6 倍,然而混合膜的质子传导率在温度升高到 100 ℃ 后开始大大降低,这说明磺酸基对质子传导的贡献很大程度上依赖水。但即使是在完全干燥的条件下,共混膜的质子传导率仍始终高于纯 PBI 膜,且共混膜比纯 ABPBI 膜有更好的热稳定性。
(2)PBI 与其他聚合物的共混
咪唑环中质子受体(—N  )的存在,使得 PBI 非常容易吸水,阻止磷酸分子与 PBI 骨架的有效接触。Jana等[81]发现,PVDF 与 PBI 在一定比例内共混能提高磷酸掺杂水平,原因是 PVDF 作为疏水性高分子,可以抑制材料的吸水能力,从而负载更多酸。
)的存在,使得 PBI 非常容易吸水,阻止磷酸分子与 PBI 骨架的有效接触。Jana等[81]发现,PVDF 与 PBI 在一定比例内共混能提高磷酸掺杂水平,原因是 PVDF 作为疏水性高分子,可以抑制材料的吸水能力,从而负载更多酸。
)的存在,使得 PBI 非常容易吸水,阻止磷酸分子与 PBI 骨架的有效接触。Jana等[81]发现,PVDF 与 PBI 在一定比例内共混能提高磷酸掺杂水平,原因是 PVDF 作为疏水性高分子,可以抑制材料的吸水能力,从而负载更多酸。为了解决磷酸流失的问题,也有研究把含膦酸基的聚合物与 PBI 共混。例如 Bozkurt 等[82]制备的 ABPBI-PVPA 混合膜。为了达到良好的混合效果,先溶解 ABPBI 于 TFA 中,然后滴加水直至溶液变得均一,再与 PVPA/水溶液混合,一系列共混膜中 ABPBI/PVPA 重复单元的摩尔比控制在 1∶0.5 至 1∶4。可惜膜的质子传导率很低,最高只有 10-3S·cm-1量级。
4.2 非 PBI 类高温质子交换膜
上文所总结的 PBI 类质子交换膜的改性手段,同理在非 PBI 类质子交换膜中也适用。因此这一部分只以高分子基材本身作为分类介绍的标准。
除 PBI 以外,磷酸掺杂的碱性聚芳醚也在高温质子交换膜中占有很大比重。其他聚合物如聚酰亚胺,又如骨架中含有碱性 N 杂环的聚乙烯吡咯烷酮、聚苯基喹喔啉、聚吡咙等高分子,都存在用于磷酸基质子交换膜的潜力,但相关研究还非常少。
4.2.1 聚芳醚类高分子
4.2.2 聚酰亚胺
4.2.3 聚乙烯吡咯烷酮/聚苯基喹喔啉
聚乙烯吡咯烷酮(Poly vinyl pyrrolidone, PVP)分子链中含有氮杂环,对于磷酸掺杂和质子传导有利,但由于水溶性,其本身不能用作质子交换膜,需要其他聚合物共混来提高机械性能,如与 PES 共混、与 PVDF 共混[86]。
He 等[87]利用聚乙烯吡咯烷酮-聚醚砜(PVP-PES)的共混物作为基材,碳量子点(CDs)作为填料,制备了一系列复合膜。原始的 PVP-PES 膜在 180 ℃ 下质子传导率为 0.029 S·cm-1,同样条件下复合膜质子传导率最高达 0.086 S·cm-1,同时可以保持溶胀率基本不变,断裂拉伸应力保持在同等的较高水平(>10 MPa)。
Lu 等[88]将 2D 结构的氮化碳(CN)纳米片用于 PVP-PES 共混物的掺杂。CN 自身吸附磷酸,同时可以提高机械性能,降低尺寸溶胀。在掺杂量达到 0.5 wt% 时,磷酸吸附量与质子传导率达到最高值。P/CN-0.5/PA 膜在 160 ℃ 下最高质子传导率达 0.104 S·cm-1,峰值功率密度达 512 mW·cm-2。
聚苯基喹喔啉(Poly phenyl quinoxaline, PPQ)具有优异的热稳定性和机械性能,且主链含有碱性N原子,具有应用于磷酸基高温质子交换膜的潜力。Hsu 等合成的 PPQ 在高浓度磷酸的掺杂过程中会发生溶解,导致尺寸稳定性和机械性能严重下降。因此他们又合成了交联的ABPPQ膜[89],大大提高了机械性能,可进行膜电极的组装,在燃料电池运行条件中表现稳定。交联膜在 160 ℃ 下最高质子传导率达 0.025 S·cm-1。
5 新型多孔材料在高温质子交换膜中的应用
5.1 多孔PBI类质子交换膜材料
Mehdipourghazi 等[99]将致孔剂 LiCl 及木质素与 PBI 混合获得了多孔膜。LiCl 导致微孔的形成,木质素是一种阴离子型聚合物,与 PBI 的微观相分离作用,能提高孔洞的数量和大小,从而能吸附更多的磷酸。多孔膜在 160 ℃ 下最高质子传导率达 0.152 S·cm-1。Xu 等[100]将 PBI 与高离子交换容量的磺化氧化石墨烯(RGO)复合,并包覆在多孔 PBI 的两侧,形成三明治结构。RGO 的引入并未对磷酸掺杂水平造成影响(可能是表面含氧基团与磷酸形成氢键),且磷酸掺杂量随着多孔层 PPBI 的质量分数提高而提高。三明治膜 PBI-RGO/PPBI-80/PBI-RGO 吸附了 500 wt% 的磷酸,ADL=20.4,在 170 ℃ 未加湿的情况下质子传导率达到 0.113 S·cm-1,拉伸强度为 22.7 MPa。
5.2 金属有机框架材料(MOFs)
金属有机框架(Metal organic frameworks, MOFs)又称多孔配位聚合物(Porous coordination polymers, PCPs),是由金属离子或团簇和有机配体之间通过配位作用连接,自组装构筑的一维、二维或三维的网络结构。MOFs 种类繁多,具有高的比表面积、可设计剪裁的框架和孔结构、较高的稳定性。过去的十年里研究 MOFs 的论文数量呈指数增长[101],相关领域包括气体储存和分离[102]、光学材料[103]、磁性材料[104]、催化[105]、质子传导[106]等。目前已有大量的 MOFs 被证实具有传导离子的潜能。制备质子传导功能的 MOFs 通常有两种策略,一种是在 MOFs 骨架上修饰磺酸基团、羧酸基团、膦酸基团等酸性官能团[107~111];另一种是从客体分子出发,在孔道中嵌入非挥发性的客体分子,例如咪唑、三氮唑、组胺、酸分子[112~114]等,提供额外的质子传导路径。MOFs 与柔性聚合物基底相混,可制备质子传导性的混合基质膜(Mixed-matrix membranes,MMM),但是迄今为止用于 MMMs 填料的 MOFs 还较少。
5.2.1 质子传导 MOFs
适合高温运行的 MOFs 目前较少。2011年,Kitagawa等[113]合成了担载组胺和咪唑分子的 MOFs,分别命名为 imidazole@Al-MOF 和 histamine@Al-MOF,后者在 150 ℃ 无水条件下的质子传导率达到 10-3 S·cm-1数量级。
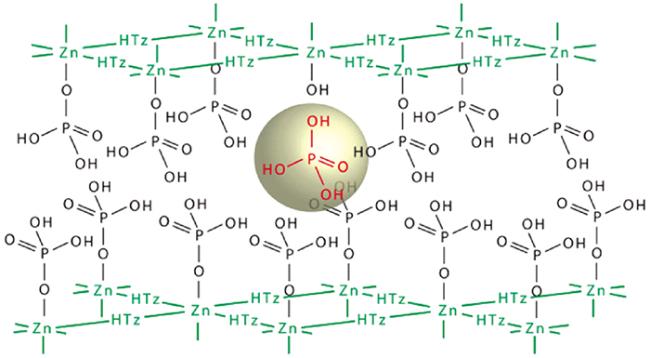
5.2.2 MOFs 复合膜
MOFs 作为固态质子传导材料,其质子传导性能多数需要压制成片测试,为满足成膜需要,与聚合物基质进行复合是一种比较简便的方法。MOFs 作为填料,多数仍是与 Nafion 或 SPEEK 复合。例如 Jiang 等[119]将浸渍植酸的 MIL-101 与 Nafion 复合,12 wt% MIL@phytic/Nafion 复合膜在 100 ℃、100% RH 条件下质子传导率可达0.228 S·cm-1,表现出比 Nafion 膜更高的性能。Zang 等[120]合成了 MIL-101、磺化 MIL-101 及 H2SO4、H3PO4、CF3SO3H 负载的 MIL-101 并用于与壳聚糖复合,负载 H2SO4、H3PO4及 CF3SO3H的复合膜在 80 ℃、35% RH 条件下最高质子传导率分别为 9.5×10-2S·cm-1、8.3×10-2S·cm-1及 9.4×10-2S·cm-1,接近 Nafion 完全水合的水平。
MOFs 与 PBI 相容性很好,尤其是含有咪唑酯结构的 ZIFs 系列。近 5 年的一些研究将 MOFs/PBI 复合膜用于气体分离[121~123],但关于 MOFs/PBI 复合质子交换膜的相关研究少之又少。2018 年 Escorihuela 等[101]通过溶液法,将沸石咪唑酯骨架材料 ZIF-8,ZIF-67 及 ZIF-8、ZIF-67 的二元混合物用于 m-PBI,得到一系列基于 PBI 的磷酸掺杂复合膜。膜的质子传导性比纯 PBI 与纯 ZIFs 都更高,含 5 wt% ZIFs的复合膜 PBI@ZIF-8、PBI@ZIF-67 在 200 ℃ 无水条件下的质子传导率分别达到 3.1×10-3 S·cm-1及 4.1×10-2 S·cm-1,PBI@ZIF-mix 的质子传导率更高达 9.2×10-2 S·cm-1,显示出 ZIFs 的协同作用。
5.3 共价有机框架材料(COFs)
COFs 有序的孔洞有利于质子的移动,且可以负载质子载体,因此大量的研究将质子载体如磷酸[127~129]、多金属氧酸盐[130]、植酸[131]、乙酸[126]等嵌入到 COFs 的孔洞内;另一种策略是直接用传导质子的基团修饰 COFs 的框架[132]。与无机填料相比,COFs 与聚合物具有更好的相容性,因此也可以用于制备混合基质膜。Banerjee 等[133]用氨基对甲苯磺酸(PTSA·H2O)盐介导,合成了一种自支撑的、高结晶度的柔性 COF,缓慢结晶过程使得质子载体 PTSA·H2O 在 COFs 的有序纳米孔内原位浸渍。负载了磺酸的 COFs 膜具有 0.078 S·cm-1 的质子传导率(80 ℃、95% RH)。Wu 等[132]合成了两性离子修饰的共价有机框架 Z-COF,Z-COF 的框架上同时含有季铵离子与磺酸基团(如图 16 所示),与 Nafion 共混得到的复合膜最高的质子传导率达 0.22 S·cm-1(80 ℃, 100% RH),比 Nafion 高出 57.1%。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
就目前的研究现状来看,无论是质子传导的 COFs 还是与其他柔性聚合物的混合膜,尽管有些已经达到较高的质子传导率,但都是在低温和较高湿度下,不能用于 HT-PEMFCs。Kharul 等[134]合成了 COF 与 PBI 的柔性共混膜并用于气体分离,由于 COFs 与聚合物相容性良好,因此用于质子交换膜具有较大的发展空间。
6 结论与展望
20 世纪 90 年代 HT-PEMFCs 的概念提出以来,高温质子交换膜已取得了丰富的研究进展。无机磷酸的热稳定性好、蒸气压低,且在高温无水条件下可自电离,因而基于磷酸基的高温质子交换膜在高运行温度、低相对湿度甚至无水条件下具有高的质子传导率、良好的化学稳定性以及热稳定性,成为最适合用于 HT-PEMFCs 的膜材料。
为提高磷酸膜的综合性能,多种改性手段已用于以聚苯并咪唑、聚芳醚等高分子为基材的高温 PEMs 中。各种改性方法的优缺点可简单总结如下:(1)由于交联剂和无机填料的选择性大,因此交联和有机-无机复合是研究中的热点,往往能够同时提高磷酸膜的质子传导率与机械性能;(2)多种聚合物复合是最为简单的改性方法,且适用性广,PBI 与磺化聚合物共混时,除了考虑相容性问题,还需考虑后者的比例越高,质子传导对湿度的依赖越大;(3)在聚合物分子链上接枝碱性基团的方法,通常只能提高磷酸掺杂率,而对机械性能的提升没有帮助;(4)与无机磷酸相比,有机膦酸基团直接以 C-P 键与高分子的主链或侧链相连,克服了无机磷酸腐蚀性强、容易流失的缺点[135],但这类质子交换膜的质子传导率一般较低,这是由于自由磷酸的数目对质子传导率的提高来说十分必要[26]。
MOFs 和 COFs 具有高的比表面积和良好的化学稳定性、热稳定性,且种类繁多、可设计性强,自身可作为晶态质子传导材料,但由于不溶、不熔的特性,制备自支撑的、具有足够力学性能的膜仍有难度,而且存在质子载体流失、燃料渗透等问题[117]。与柔性聚合物复合似乎能容易地满足燃料电池质子交换膜的需要。
高温燃料电池的理论与实验研究仍在快速发展,磷酸掺杂型高温燃料电池质子交换膜的研究思路也将不断拓宽,如何降低成本、延长工作寿命将成为 PEMs 商用所面临的重要课题。