




1 引言
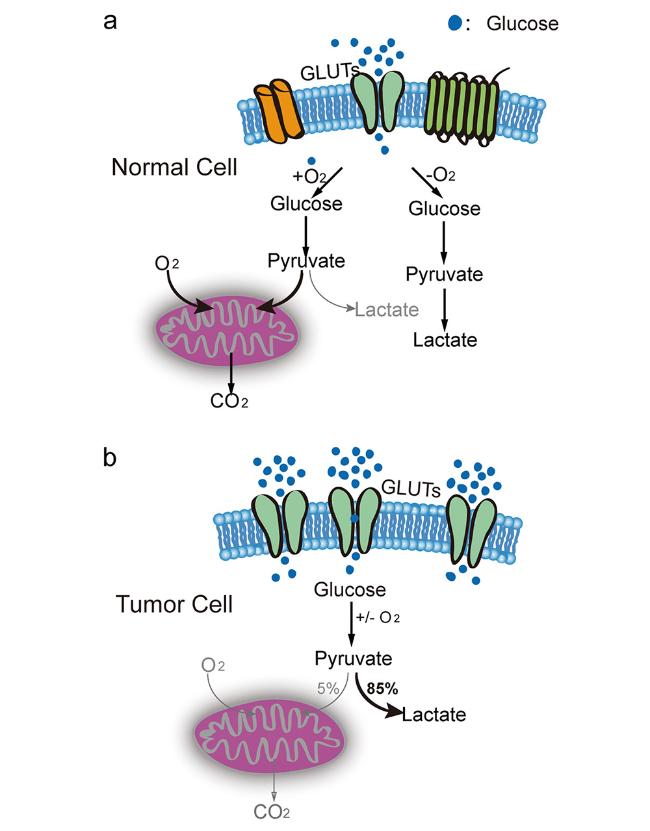
1924年,德国生物化学家Otto Warburg等揭示了肿瘤细胞以一种不同于正常细胞的葡萄糖代谢途径产生能量维持其生存和增殖[1]。在有氧的环境中,多数正常细胞是依靠线粒体的三羧酸循环(TCA cycle,tricarboxylic acid cycle)以氧化途径代谢葡萄糖分子产生还原性辅酶NADH、二氧化碳和水,然后经过氧化磷酸化(OXPHOS,oxidative phosphorylation)产生大量的ATP和非常少量的乳酸(如 图1a)。只有在缺氧或低氧环境中,分化的细胞才会产生大量的乳酸和少量的ATP[2]。然而,大多数肿瘤细胞即使在氧气充足的环境中,也是通过产能效率相对较低的糖酵解作用(glycolysis)为自身供能(如图1b),这就是著名的沃伯格效应(Warburg Effect)[3]。肿瘤细胞为了维持自身快速生长、增殖和转移的需要,必须大量表达葡萄糖转运蛋白GLUTs(glucose transporters)才能满足其对葡萄糖的摄取和代谢需求,因此,过量表达GLUTs成为肿瘤细胞的重要标识之一,也促使GLUTs成为开发抗肿瘤药物的重要研究靶点。值得一提的是,自人类发现糖分子转运蛋白GLUT以来时隔四十年,我国科学家清华大学医学院颜宁于2014和2015年首次成功解析了GLUT1和GLUT3的晶体结构。该成果对研究肿瘤高表达GLUTs的“沃伯格效应”重新赋予了结构生物学基础[4,5]。
2 葡萄糖转运蛋白
葡萄糖转运蛋白是人体内最重要的跨膜蛋白之一,共有14种亚型(GLUT1-GLUT14),目前针对GLUT1-GLUT4的生理学机制研究的最为清楚,它们分别负责人体不同组织器官中的葡萄糖转运和再吸收。其中,GLUT1是分布最广泛、研究最多的葡萄糖转运蛋白,它由SLC2A1基因编码,主要负责葡萄糖进入红细胞以及跨越血脑屏障,因此,在脑部、红细胞、血脑屏障、骨骼肌和脂肪组织中表达量较高[6]。2014年,Yan等[4]解析了分辨率为3.2 Å的向内开放的人类GLUT1晶体结构,其蛋白结构由12个跨膜螺旋组成了N端和C端两个结构域,两个结构域之间的腔孔朝向胞内,这种构象是协同转运蛋白超家族(major facilitator superfamily,MFS)的一种经典结构。利用GLUT1的蛋白晶体结构,研究人员精确分析了与疾病相关的三十多个氨基酸突变位点,这些突变主要集中在底物结合口袋、胞外门控区和胞内门控区,并详细介绍了相关疾病的致病机理。进一步研究发现,GLUT1的过量表达与多种肿瘤的类型和能量代谢密切相关。早前Grover-McKay等[7]报道了GLUT1的过度表达与乳腺癌细胞的侵袭能力有关,乳腺癌细胞会随着侵袭力的增强表达更多的GLUT1。Cruz等[8]对81例肠镜检查患者进行直肠活检时发现在结肠处有癌前病变的患者GLUT1表达量显著提高。Alves等[9]发现GLUT1在原发性肝癌细胞中的表达高于正常肝细胞,同时其表达量随着肝癌恶化而增加并直接影响肝癌的预后。Yan等[10]对胃癌患者体内癌细胞的研究证实了SLC2A1基因过度表达与胃癌细胞的增殖和转移成正相关。除此之外,GLUT1的高表达与非小细胞肺癌[11]、前列腺癌[12]、口腔鳞癌[13]、卵巢癌[14]、膀胱癌[15]、胰腺癌[16]、黑色素瘤[17]等多种恶性肿瘤的发生和预后息息相关,这使得GLUT1的表达量成为细胞癌变的重要临床检测指标之一,通过靶向GLUT1开发抗肿瘤药物或诊断试剂,阻断细胞的营养来源或将药物伪装成营养物质大量供给到肿瘤细胞内,成为抗肿瘤试剂研发的重要策略。
GLUT2由SLC2A2基因编码,是肝脏、胰腺、肠道、肾脏和大脑中主要的葡萄糖转运体,除葡萄糖的运送和传递之外,对半乳糖、甘露糖和果糖也具有低亲合力和广泛的底物特异性。同时,GLUT2的表达与葡萄糖浓度、胰岛素的分泌、自主神经系统的活动以及进食和体温调节等有关[18]。因此,GLUT2与糖尿病[19]和自身免疫性糖尿病[20]之间存在密切关联。此外,研究人员在肝癌[21]、乳腺癌[22]和胃癌[23]等少数肿瘤中也检测到了高表达的GLUT2。GLUT3多表达在神经细胞和血小板中,在非小细胞肺癌[24]、膀胱癌[25]、口腔鳞状细胞癌[26]、喉癌[27]等多种实体瘤中GLUT3也均高表达。GLUT4是胰岛素敏感的葡萄糖转运载体,主要存在于骨骼肌、心肌及脂肪组织中,主要作用于调节葡萄糖吸收和转运以适应葡萄糖代谢增强的需求。2-型糖尿病、肥胖和衰老等与GLUT4的表达和功能受损紧密相关[28]。综上,目前的研究表明GLUT1和GLUT3与多种实体瘤和恶性肿瘤的发生与发展关系密切,被认为是靶向肿瘤沃伯格效应的特异性生物标志物。
3 靶向沃伯格效应抗癌药物的研究现状
过量表达GLUTs作为沃伯格效应的特异性生物标识,开发能够与其特异性结合的抑制剂,理论上可阻断肿瘤细胞的葡萄糖摄取,切断肿瘤的营养供应途径,因此是肿瘤诊断试剂及治疗药物开发的一种思路。然而,不同亚型的GLUT在大多数哺乳动物细胞中广泛表达,如何选择性阻断肿瘤细胞的葡萄糖摄取是现阶段GLUTs抑制剂类药物开发的研究重点。其次,根据肿瘤细胞葡萄糖代谢亢进的生理特征,将GLUTs底物与药物分子有机结合形成糖偶联化合物,使药物作为肿瘤细胞的代谢营养被肿瘤组织主动摄取,从而达到靶向抗肿瘤作用也是一种行之有效的靶向沃伯格效应抗肿瘤策略。
3.1 GLUTs抑制剂
3.1.1 天然产物类抑制剂
GLUTs抑制剂中有许多是天然产物(如图2)。根皮素(Phloretin)是第一个被发现的黄酮类GLUT1抑制剂,同时也是GLUT2抑制剂,可以与红细胞可逆结合[29],因此能够选择性抑制肿瘤细胞生长,正处于临床前研究阶段。细胞松弛素B(Cytochalasin B)是一类抑制肌动蛋白的真菌代谢物,是对GLUT1具有较高特异性的抑制剂。除有效抑制糖吸收之外它还能通过破坏肌动蛋白调控的电压门控钾离子通道(Kv1.5)直接干扰肿瘤细胞的增殖和转移[30]。此外,细胞松弛素B也可作为一种开放通道阻滞剂,以一种独立于细胞骨架的方式直接阻断Kv1.5[31],影响癌细胞的生长[32]。白藜芦醇(Resveratrol)也是一种天然的GLUT1抑制剂,
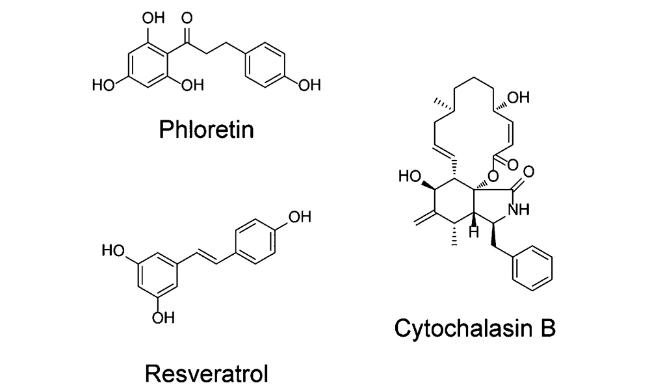
具有抑瘤、抗炎及降糖功效。白藜芦醇除了能够抑制细胞吸收葡萄糖从而影响糖代谢以外,还可通过下调NF-κB(nuclear factor-kappa B)信号级联和上皮细胞间质转化(epithelial-to-mesenchymal transition,EMT)介导肿瘤坏死因子受体相关因子6(tumor necrosis factor receptor associated factor 6,TRAF6)的溶酶体降解,从而抑制前列腺癌细胞生长[33]。2017年,Resveratrol被美国FDA批准为脊髓小脑性共济失调(spinocerebellar ataxia)和弗里德希氏共济失调(friedreich’s ataxia)综合症的孤儿药。同时,Resveratrol已经进入治疗2型糖尿病的临床二期试验研究[34]。另外,许多多酚类物质也被证明是GLUTs抑制剂,它们多存在于植物和水果蔬菜中,对多种癌症都有抑制作用。例如,苹果多酚素(Apple polyphenol phloretin)是一种GLUT2的特异性抑制剂,能够抑制结直肠癌细胞的侵袭[35]和人三阴性乳腺癌细胞的增殖和扩散[36]。除此之外,橙皮素、柚皮素、儿茶素、棉酚、姜黄素、豆蔻明、槲皮素和槲皮苷等也均属于天然产物类GLUTs抑制剂[37]。然而,Phloretin和Cytochalasin B等天然产物类GLUT抑制剂由于选择性差和不可成药性等问题,大大限制了其临床应用。
3.1.2 处于临床前体外研究阶段的GLUTs抑制剂
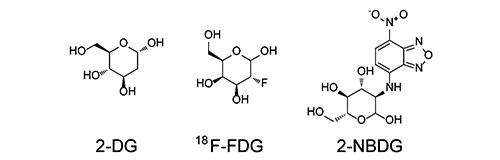
目前,许多具有抗肿瘤活性的GLUTs小分子抑制剂正处于临床前开发阶段(如图3)。2008年,Yang等[38]发现噻唑烷二酮过氧化物酶增殖物激活受体γ(peroxisome proliferator-activated receptor γ,PPARγ)激动剂可以有效阻止葡萄糖的吸收,具有良好的抗肿瘤活性。随后,Wang等[39]在这个受体激动剂的基础上进行结构衍生与修饰,设计并合成了30个化合物,经过针对前列腺癌细胞(LNCaP细胞)的体外抗肿瘤活性测试,发现化合物1对LNCaP细胞表现出更强的葡萄糖摄取抑制作用(IC50=2.5 μM),且对正常前列腺上皮细胞和正常乳腺上皮细胞没有产生明显的毒性。通过分子对接分析发现化合物1对葡萄糖的抑制作用归因于与GLUT1的选择性结合,因此被认为是一种有效的GLUT1抑制剂。2016年,Ung等[40]基于大肠杆菌木糖转运蛋白( E. coli xylose transporter,XyIE)的X射线晶体结构,构建了人GLUT1(human GLUT1,hGLUT1)同源模型。通过与已确定的hGLUT1晶体结构对比,发现了葡萄糖结合位点旁边存在一个疏水口袋。计算机辅助虚拟筛选研究最终发现了7种新的GLUT1配体(IC50=0.45~59 μM),并利用高表达hGLUT1的中国仓鼠乳腺细胞(Chinese hamster ovary,CHO)进行2-脱氧-D-葡萄糖(2-deoxy-D-glucose,2-DG)吸收实验,发现化合物PUG-1作为GLUT1抑制剂能显著抑制2-DG的吸收(IC50=0.45 μM)。Granchi等[41]利用四芳基取代的水杨酮肟结构骨架合成了一系列对肺癌细胞有明显抑制作用的化合物,其中,化合物2抑制肺癌细胞(A549)的活性最好(IC50=7.0 μM),优于同类型的其他GLUT1抑制剂。随后通过荧光显微镜观察到,化合物2处理后的A549癌细胞对GLUT1荧光底物2-( N-7-硝基-2,1,3-苯并口恶二唑-4-氨基)-2-脱氧-D-葡萄糖(2-( N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-2-deoxyglucose,2-NBDG)的吸收明显减少(下降约38%)。2018年,Karageorgis等[42]将色原烷和四氢嘧啶酮进行生物定向合成(biology-oriented synthesis,BIOS),制备了一种新型靶向GLUT1和GLUT3的抑制剂Chromopynone-1,并发现它能有效抑制胰腺癌细胞(MIA PaCa-2)和结肠癌细胞(HCT116)的生长。2019年,Ceballos等[43]也基于吲哚胺类天然产物的母核结构,衍生合成了一种靶向GLUT1和GLUT3的抑制剂Glupin,证实能够有效抑制乳腺癌细胞生长。
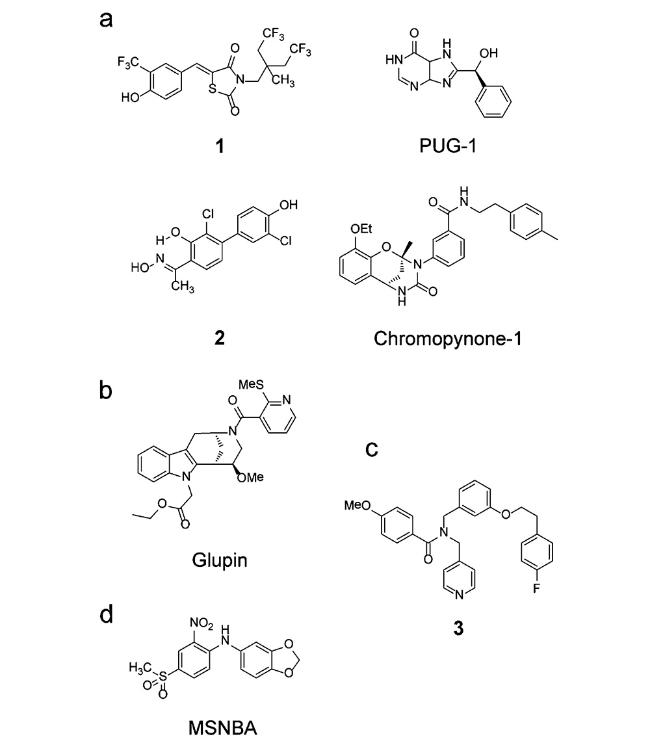
Wei等[44]利用GLUT4同源模型与小分子化合物进行高通量虚拟筛选,发现其中某些化合物能够与GLUT4的关键残基(Asn176和Ile42)相互作用,同时对其他GLUT亚型具有选择性。随后,研究人员将与GLUT4亲合力最高的化合物进行结构修饰,设计并合成了16个衍生物,其中化合物3对人多发性骨髓瘤细胞(KMS11)的增殖抑制作用最强(IC50=13.41 μM)。通过将抑制剂与人工构建的GLUT1、GLUT2、GLUT3、GLUT4、GLUT8高表达人胚胎肾细胞(HKE293)株进行葡萄糖转运抑制作用研究,发现化合物3对GLUT4的选择性明显高于GLUT1、2、3和8,进一步证明了化合物3是一种GLUT4介导的葡萄糖转运抑制剂(IC50=18.2 μM)。随后的人白血病细胞(JJN3)和多发性骨髓瘤细胞(L363)抑制结果显示,与JJN3细胞相比,化合物3能够显著降低L363细胞中促生存抗凋亡蛋白(myeloid cell leukemia 1,MCL-1)的表达,从而有效抑制L363的生长。
MSNBA是Thompson等[45]利用GLUT5模型筛选的首个GLUT5抑制剂。在人乳腺癌细胞系(MCF-7)中,MSNBA能竞争性抑制GLUT5摄取果糖( K i=(3.2 ± 0.4) μM),且不影响GLUT2的果糖转运和GLUT1-4的葡萄糖转运。通过分子对接分析发现,MSNBA结合在GLUT5的H387活性位点附近。
3.1.3 处于临床前体内研究阶段的小分子抑制剂
许多靶向GLUTs的小分子抑制剂在临床前体内药效研究中都展示出良好的生物学活性(如图4)。2012年,Liu等[46]研究合成了具有GLUT1、GLUT3和GLUT4抑制活性的多酚类化合物WZB117。该化合物主要作为GLUT1抑制剂:在肺癌细胞(A549)的2-DG竞争性吸收实验中,WZB117能够显著抑制肿瘤细胞摄取2-DG;在荷瘤裸鼠模型药效试验中,与空白对照组相比,实验组小鼠每天腹腔注射该抑制剂,给药10周后肿瘤体积减少了约70%。该研究系列中的WZB27和WZB115也是很好的GLUT1抑制剂,它们能够通过抑制葡萄糖吸收来抑制癌细胞生长并诱导细胞凋亡。与普通细胞相比,WZB27和WZB115还能明显抑制人非小细胞肺癌细胞(H1299和A549)和MCF-7细胞的生长。当WZB27或WZB115与抗癌药物顺铂或紫杉醇联合使用时,能够协同抑制癌细胞的生长[47]。
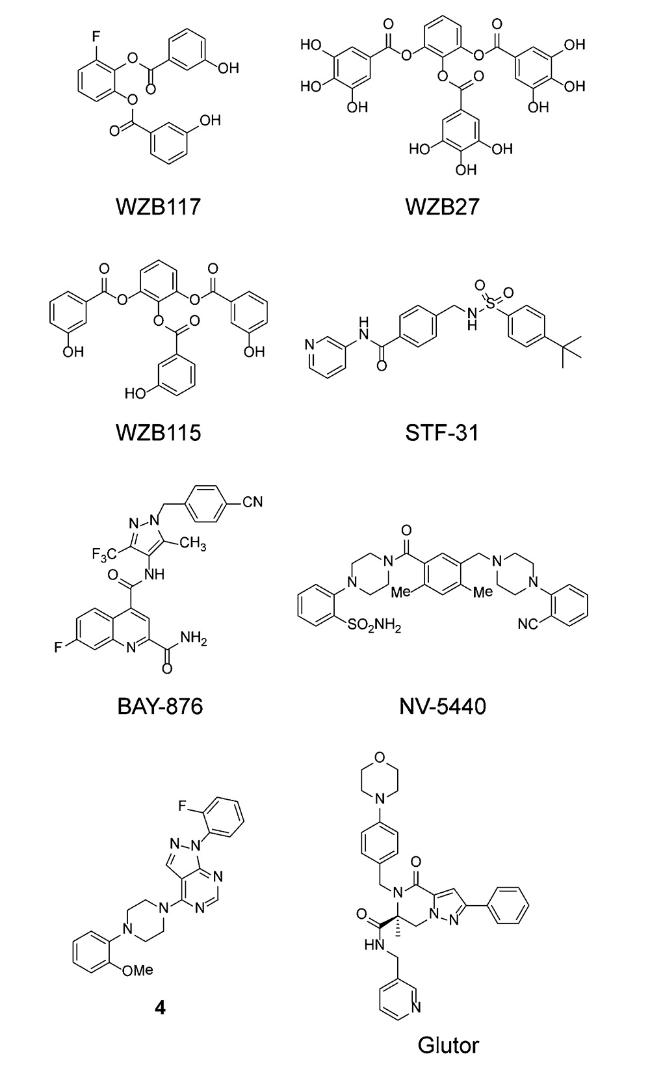
希佩尔-林道综合症(von Hippel-Lindau,VHL)是由于VHL抑癌基因缺失导致的,大约80%的肾细胞癌中都会发生这种基因缺失。为了研究一种选择性靶向肾细胞癌的药物,Chan等[48]利用高通量化学合成和药效筛选发现了以STF-31为代表的一系列对VHL基因缺失肾癌具有抑制作用的小分子。研究人员通过分子对接,证明了STF-31与GLUT1结合的特异性,并在体内外实验中验证了STF-31能够抑制GLUT1荧光底物2-NBDG的吸收以及诱导肾癌细胞凋亡等药效机制。Ma等[49]研究的BAY-876是一种以 N-(1 H-吡唑啉-4-氰基)喹啉-4-羧胺为骨架的GLUT1抑制剂,该抑制剂具有良好的选择性,能够有效阻断肿瘤细胞的糖酵解途径,从而抑制人卵巢癌(SKOV-3)细胞的增殖(IC50=60 nM)。在SKOV-3小鼠异种移植模型中,BAY-876每日给药剂量4.5 mg/kg,连续给药28~30 d后,肿瘤体积和重量平均减少68%。体内药代动力学研究表明,BAY-876在Wistar大鼠( F%=85%, T 1/2=2.5 h)和Beagle犬( F%=79%, T 1/2=22 h)中有良好的口服生物利用度和较长的末端半衰期,并且体外药代动力学数据也显示BAY-876在不同被测物种(人、猴、犬和大鼠)的肝微粒体和肝细胞中高度稳定[50]。
雷帕霉素靶蛋白(mechanistic target of rapamycin,mTOR)是哺乳动物细胞代谢的中心调控因子,是两个大分子信号复合物mTORC1和mTORC2的催化亚基,其中mTORC1在调节细胞生长以及细胞糖代谢方面起着关键作用。Kang等[51]在寻找mTORC1抑制剂时发现,化合物NV-5440与其他四种类似物相比活性最强,能够通过阻断葡萄糖摄取(IC50=36 nM)导致mTORC1信号失调。通过荧光交叉相关光谱(fluorescence cross-correlation spectroscopy,FCCS)实验,确认了NV-5440能直接与GLUT1结合。利用MCF-7细胞进行代谢通量分析时,研究人员还发现NV-5440可与包括GLUT1在内的多个调节位点相互作用,从而有效抑制糖酵解过程。
Siebeneicher等[52]通过高通量筛选发现具有1H-吡唑并[3,4 -d]嘧啶母核结构的化合物是一类有效的GLUT1抑制剂。该类化合物左侧芳环上的邻位甲氧基(—OCH 3)使其对GLUT1有高亲合性并对GLUT2有良好的选择性。化合物中心的哌嗪环对抑制GLUT1活性起决定作用。因此,以1 H-吡唑并[3,4 -d]嘧啶为母核,科研人员设计并合成了一系列衍生物。利用高表达hGLUT1的DLD1细胞、高表达hGLUT2的CHO细胞以及敲除hGLUT1后主要表达hGLUT3的DLD1细胞,发现化合物4对GLUT1细胞表现出很强的抑制效果(IC 50=7 nM),同时对GLUT2和GLUT3高表达细胞也具有一定的选择性(GLUT2和GLUT3的IC50分别为1.1 μM和40 nM)。体外药代动力学实验发现,化合物4在Wistar大鼠体内的血液清除率较低(占肝脏血流量的29%),口服生物利用度高达67%。
Glutor是一种基于哌嗪的GLUT1、GLUT2和GLUT3广谱抑制剂,能够有效抑制癌细胞摄取2-DG(IC50=11 nM)。研究发现,在2D和3D的癌细胞培养中,Glutor能够有效抑制多种癌细胞生长,且对GLUT3具有更好的选择性(优于GLUT1)。将Glutor和谷氨酰胺酶抑制剂CB-839联合使用,能协同抑制结肠癌细胞(HCT116)生长[53]。
3.2 葡萄糖类似物与糖偶联化合物
基于沃伯格效应,肿瘤细胞较正常细胞相比过度表达GLUTs并大量摄取葡萄糖,以满足自身的代谢需求,因此,开发具有抗肿瘤效果的葡萄糖类似物或将药物分子与糖分子有机结合成糖偶联化合物,通过靶向沃伯格效应抑制肿瘤细胞的生长成为一种新的抗肿瘤药物开发思路。
3.2.1 葡萄糖类似物
| Phases | Combined therapy | Diseases | Status |
|---|---|---|---|
| Phase Ⅲ | — | Prostate cancer | Terminated(Has results) |
| Phase Ⅰ | Docetaxel | Lung/Breast/Head and neck cancer | Completed |
| Phase Ⅰ | Radiotherapy | Glioblastoma multiforme | — |
| Phase Ⅲ | Radiotherapy | Cerebal gliomas | — |
3.2.2 糖偶联化合物
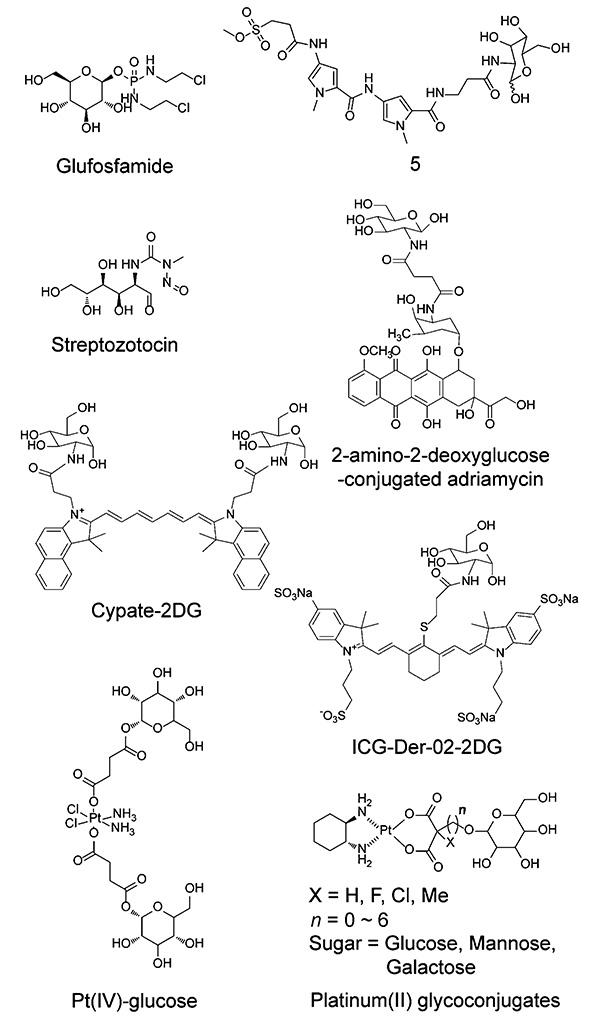
近年来,通过糖分子修饰或糖偶联策略开发肿瘤治疗药物的研究越来越多(如图6),尤其是针对顺铂、卡铂、奥沙利铂等铂类化合物以及其他毒性较高溶解性较差的抗肿瘤药物。1995年,Wiessier等[61]合成了第一个具有广谱抗肿瘤活性的糖偶联药物——葡磷酰胺(Glufosfamide)。它是由一分子葡萄糖与一分子具有DNA烷化作用的异磷酰胺氮芥通过糖苷键偶联形成的一种前药。该前体药物通过GLUT1的转运进入肿瘤细胞后,发生水解释放药效成分异磷酰胺氮芥从而发挥抑癌作用[62]。与异环磷酰胺相比,葡磷酰胺实现了药物对肿瘤细胞的靶向性,水溶性大大提高,毒副作用显著降低[63]。DNA烷基化药物也是治疗肿瘤的重要武器,然而由于该类化合物缺乏靶向性,因此在杀死肿瘤细胞的同时也对正常组织细胞造成极大损伤。目前,葡磷酰胺正处于转移性胰腺癌和晚期乳腺癌患者的临床3期试验研究中。同时,葡磷酰胺与氟尿嘧啶(Fluorouracil,5-FU)作为二线用药,研究它们对吉西他滨一线化疗后转移性胰腺癌患者的治疗工作也正处于临床3期。Buchanan等[64]将DNA烷基化药物的一端添加葡萄糖胺单元,合成了具有良好的肿瘤细胞选择性的化合物5。链脲霉素(Streptozotocin)[65]也是临床上使用的具有靶向性的糖偶联DNA烷基化药物。此外,研究人员还将其他DNA烷基化药物如白消安(Busulfan)、多米卡星(Duocarmycin)及苯丁酸氮芥(Chlorambucil)等与葡萄糖等糖类结合,形成新的具有靶向性的抗肿瘤药物,极大地降低了对机体正常组织的毒副作用[66];通过将葡萄糖、甘露糖、半乳糖、木糖等糖类与紫杉醇、多西紫杉醇等紫杉烷类药物偶联,既提高了药物的靶向性、降低药物毒副作用,又大大增加了药物的水溶性、提高了药物本身的生物利用度[67 ⇓~ 69]。Cao等[70]将拓扑异构酶Ⅱ抑制剂阿霉素通过琥珀酸与2-氨基-2脱氧葡萄糖偶联,增强了阿霉素对肿瘤细胞的选择性。Reinhard等[71]将葡萄糖与苄基鸟嘌呤类药物(O6-甲基鸟嘌呤-DNA甲基转移酶(MGMT)抑制剂)结合,既增强了酶的抑制活性,又增加了原化合物的靶向性。Guo等[72]分别将近红外荧光燃料多次甲基菁染料(Cypate)和吲哚菁绿衍生物(ICG-Der-02)与2-DG偶联,合成了选择性高的Cypate-2DG和ICG-Der-02-2DG近红外小分子荧光探针,该探针在水溶性和肿瘤靶向性方面都得到了极大改善,且在荷瘤裸鼠模型中具有良好的肿瘤蓄积效果,因此有望被开发应用于肿瘤光学成像及葡萄糖相关疾病病理研究中。
铂类抗肿瘤药物因具有广谱性强和抗肿瘤活性高等特点,是目前临床肿瘤化疗中应用最广泛的药物之一。然而,由于该类药物水溶性低、靶向性差、毒副作用强以及容易造成耐药等问题,在临床使用中受到一定限制。天津大学药学院Gao等自2001年起致力于研究糖偶联-铂(Ⅱ)类化合物在抗肿瘤研究中的应用,通过在铂类化合物中引入糖分子,改善药物的水溶性,靶向肿瘤沃伯格效应,提高药物分子的选择性。2013年,他们将不同的糖分子与奥沙利铂类似物偶联,首次报道了不同的糖偶联铂(Ⅱ)配合物[73]。该类化合物作为GLUT1的转运底物,能够被高表达GLUT1的肿瘤细胞选择性主动吸收,并且较奥沙利铂溶解度提高约150倍。抗肿瘤药效方面,含氟糖偶联铂(Ⅱ)配合物(Platinum(Ⅱ)glyconjugates)(图6)针对人结肠癌(HT29)的细胞毒性约为奥沙利铂的10倍,在HT29荷瘤裸鼠体内药效研究中,同等毒性剂量尾静脉每周一次共四次给药研究结果显示,含氟糖偶联铂(Ⅱ)配合物不仅抑瘤效果优于临床药物奥沙利铂,且其治疗指数是奥沙利铂的30倍以上[74]。除此之外,针对Warburg效应,他们设计出一系列新的2-DG偶联铂(Ⅱ)配合物,用于GLUT1介导的抗癌药物靶向递送。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
4 结论与展望
作为人类长期以来对肿瘤研究的重要成果,沃伯格效应揭示了恶性肿瘤细胞糖代谢旺盛的特征及肿瘤细胞与正常细胞GLUTs表达量的差异,明确了GLUTs是开发抗肿瘤治疗药物关键生物靶点的重要地位。本文以GLUTs为出发点,针对其抑制剂、转运底物以及糖偶联抗肿瘤药物的设计及研究进展进行了概述,为利用肿瘤沃伯格效应设计和开发具有更高临床应用价值的新型靶向抗肿瘤药物提供了新思路。然而,目前临床上明确以GLUTs为靶点的抗肿瘤药物研究仍然匮乏,多数研究仍处于临床前研究阶段。因此,开发全新的能够精准靶向GLUTs的有效抗肿瘤药物、将传统抗肿瘤药物底物化或糖偶联靶向化,仍然是开发抗肿瘤药物的重要研究方向之一。