





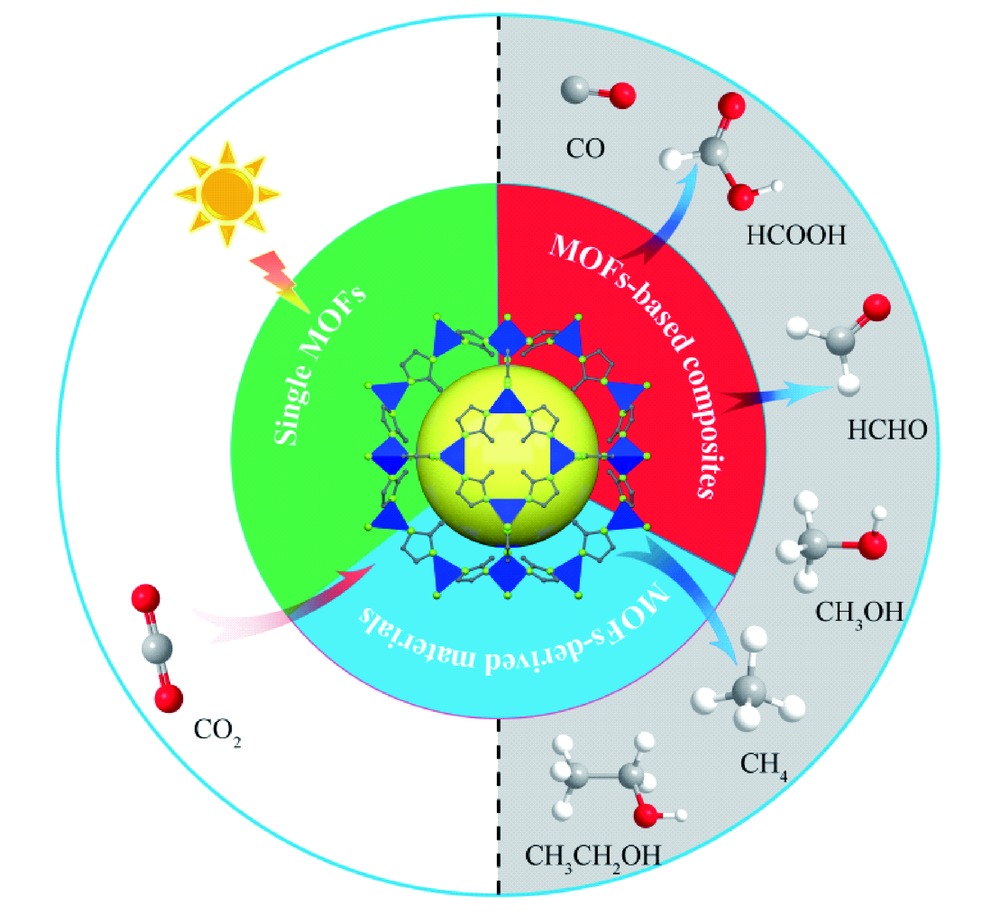
1 引言
在过去的几十年里,由于化石燃料的大量消耗,二氧化碳(CO2)的过度排放导致环境问题日益严重。如何将CO2有效地利用起来成为全世界的研究热点。与高耗能的CO2捕集和储存(CO2 capture and storage, CCS)技术相比,将CO2转化为有价值的能源燃料是一种能同时解决能源危机和环境问题的有效策略[1, 2]。到目前为止,人们已经探索了许多将CO2转化为碳氢燃料的方法,包括传统热催化、光催化、电催化和光电催化等[3]。而光催化还原CO2由于使用清洁可再生的太阳能受到越来越多的关注。CO2分子中C O双键的键能高达750 kJ·mol-1,具有很高的热力学稳定性,C
O双键的键能高达750 kJ·mol-1,具有很高的热力学稳定性,C O键的断裂需要较高能量。根据转移电子数(n=2、4、6、8、12甚至更多的电子)的不同,CO2的还原产物主要有CO、HCOOH、HCHO、CH3OH、CH4、C2H5OH等,当反应体系中有水存在时还可能生成H2。CO2在水中的部分还原过程及其氧化还原电势(vs NHE, pH=7)如反应式(1)~(8)所示[4,5,6]。要使CO2光还原半反应能够顺利进行,半导体催化剂的导带(CB)必须比CO2还原半反应的还原电势更负。
O键的断裂需要较高能量。根据转移电子数(n=2、4、6、8、12甚至更多的电子)的不同,CO2的还原产物主要有CO、HCOOH、HCHO、CH3OH、CH4、C2H5OH等,当反应体系中有水存在时还可能生成H2。CO2在水中的部分还原过程及其氧化还原电势(vs NHE, pH=7)如反应式(1)~(8)所示[4,5,6]。要使CO2光还原半反应能够顺利进行,半导体催化剂的导带(CB)必须比CO2还原半反应的还原电势更负。
O双键的键能高达750 kJ·mol-1,具有很高的热力学稳定性,C O键的断裂需要较高能量。根据转移电子数(n=2、4、6、8、12甚至更多的电子)的不同,CO2的还原产物主要有CO、HCOOH、HCHO、CH3OH、CH4、C2H5OH等,当反应体系中有水存在时还可能生成H2。CO2在水中的部分还原过程及其氧化还原电势(vs NHE, pH=7)如反应式(1)~(8)所示[4,5,6]。要使CO2光还原半反应能够顺利进行,半导体催化剂的导带(CB)必须比CO2还原半反应的还原电势更负。 CO2 + 2e- + 2H+ → HCOOH E1=-0.61 V
CO2 + 2e- + 2H+ → CO + H2O E2=-0.53 V
CO2 + 4e- + 4H+ → HCHO + H2O E3=-0.48 V
CO2 + 6e- + 6H+ → CH3OH + H2O E4=-0.38 V
CO2 + 8e- + 8H+ → CH4 + 2H2O E5=-0.24 V
2CO2 + 8H2O + 12e-→ C2H4 + 12OH- E6=-0.34 V
2CO2 + 9H2O + 12e- → C2H5OH + 12OH- E7=-0.33 V
3CO2 + 13H2O + 18e- → C3H7OH + 18OH- E8=-0.32 V
2H+ + 2e- → H2 E9=-0.41 V
2H2O + 4h+ → O2 + 4H+ E10=+0.82 V
还原产物主要取决于催化剂的种类以及催化反应的条件。近年来已经报道了许多不同种类的均相[7,8,9]和非均相[10,11,12]光催化CO2还原体系。与均相体系相比,非均相体系具有更多的优点和更广泛的应用前景[13]。光催化CO2还原反应主要分为3步:(1)光的吸收:半导体催化剂吸收能量大于等于其禁带宽度的入射光;(2)光生电荷的分离和迁移:半导体价带上的电子吸收能量后跃迁至导带,光生空穴则留在价带。随后,光生载流子迁移至催化剂表面的活性位点参与随后的氧化/还原反应;(3)氧化/还原反应:迁移至催化剂表面的电子和空穴分别参与CO2的还原反应以及牺牲试剂的氧化反应。目前,光催化CO2还原过程仍然存在很多缺点,如可见光响应能力低、光生电子空穴对复合严重、CO2吸附量小、产物的选择性低以及在含水环境中的产氢竞争反应(式9)等[14,15,16]。高效稳定的CO2光还原催化剂的设计和制备仍是未来的研究重点。
金属-有机框架(Metal-organic framework, MOFs)是一类由金属离子/簇和有机配体通过配位组装形成的有序多孔晶态材料,具有均一的孔隙结构、超大的比表面积以及灵活多变的网络拓扑结构等,已在气体的存储和分离[17]、电化学储能[18]、多相催化[19,20,21]、化学传感[22, 23]、生物医学治疗诊断[24]等诸多领域受到广泛关注。在CO2的吸附与分离中,MOFs材料凭借其较大的比表面积和CO2吸附量表现出独特的优势,而其半导体特性使得MOFs材料在光还原CO2催化反应中具有良好的应用前景[25,26,27]。但是MOFs的可见光响应能力弱,光生电子-空穴的复合比较严重,为了提高MOFs材料光还原CO2的催化性能,现有研究主要从以下三个方面入手:(1) 金属节点的改性以及有机配体的功能化修饰;(2) 半导体、贵金属纳米颗粒、钙钛矿量子点等具有光吸收能力的材料的嵌入;(3) MOFs衍生材料。本文主要对当前MOFs材料在光催化CO2还原中的发展现状、存在问题及其解决方法进行了分析和总结,最后对其未来的发展趋势进行了展望。
2 单一MOFs催化
在MOFs结构中,有机配体不仅仅起到支撑、稳定框架结构的作用,还能够在特定波长的太阳光照射下激发出电子,这使得MOFs材料成为有发展前景的半导体材料。通过对有机配体进行功能化修饰可以增强MOFs催化剂光还原CO2的催化性能。
2.1 有机配体作为光敏剂
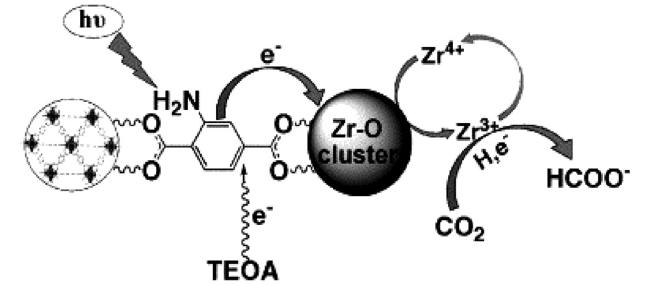
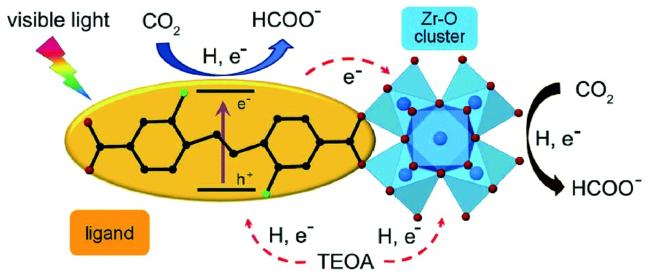
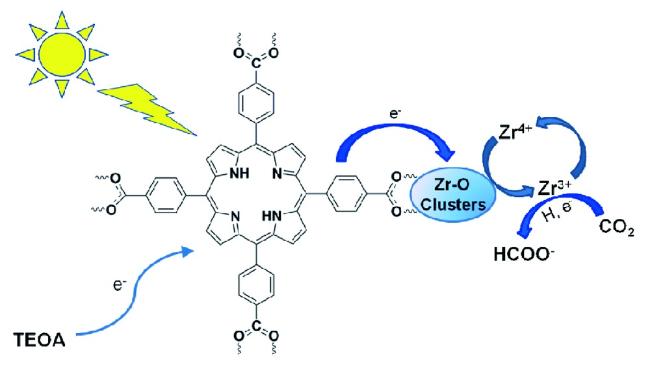
Sun等[30]以2-氨基对苯二甲酸(ATA)为配体合成了NH2-UiO-66(Zr)催化剂。在可见光照射下反应10 h,NH2-UiO-66(Zr)催化还原CO2生成HCOO-的量为13.2 μmol,而单独使用ATA配体并未表现出催化性能,说明Zr-O簇是催化反应的活性中心。通过电子自旋共振(ESR)和光致发光光谱(PL)阐明了光致电荷的迁移过程以及光催化CO2的还原过程。如图1所示,有机配体在吸收了可见光后,激发出的电子快速迁移到Zr-O簇,将ZrⅣ还原成ZrⅢ,吸附态的CO2被ZrⅢ还原成HCOO-,而ZrⅢ被重新氧化成ZrⅣ。三乙醇胺(TEOA)作为电子牺牲试剂补充消耗的电子,使催化循环能够持续进行。当使用2,5-二氨基对苯二甲酸(DTA)代替部分ATA时,含有混合配体的NH2-UiO-66(Zr)对可见光的吸收和CO2的吸附显著增强,光催化活性进一步提高了50%。
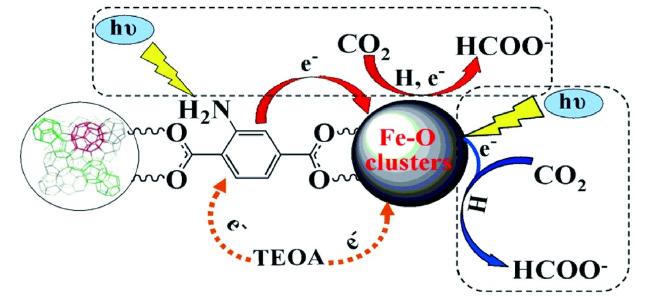
Wang等[31]研究了三种铁基MOFs(MIL-101(Fe)、MIL-53(Fe)、MIL-88B(Fe))及其氨基功能化后的光还原CO2催化性能。MIL-101(Fe)上配位不饱和的Fe金属中心能够吸附CO2,而ESR信号的变化表明Fe是光还原CO2的活性中心,因此MIL-101(Fe)表现出最佳的光催化性能。氨基功能化修饰后,Fe基MOFs材料的可见光吸收能力以及对CO2的吸附能力增强,有利于光还原CO2反应的进行。通过紫外-可见漫反射(UV-DRS)和ESR结果分析可知,氨基功能化Fe-MOFs中存在的两种激发路径是催化性能增强的主要原因,即Fe-O簇和—NH2吸光后均可激发出电子,电子分别从Fe-O簇的O2-以及有机配体迁移到Fe3+中心,通过Fe3+/Fe2+的氧化还原反应将CO2还原成HCOO-(图2)。同样,TEOA作为电子牺牲试剂补充消耗的电子,使催化循环能够持续进行。
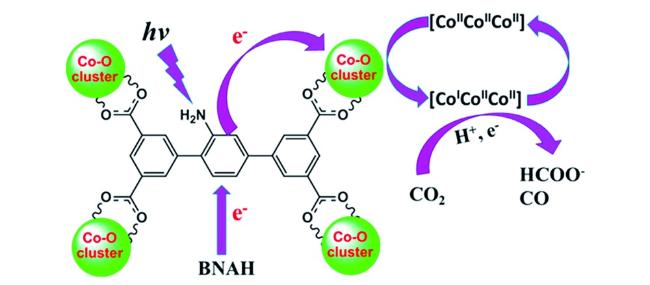
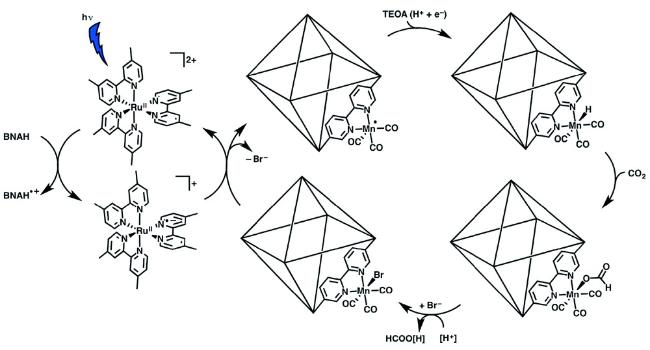
Liao等[32]采用一锅溶剂热法成功合成了一种多功能Co-MOF [Co3(HL)2·4DMF·4H2O]光催化剂,同时表现出较高的产氢和光还原CO2催化活性。不需要额外添加光敏剂和助催化剂,Co-MOF在可见光下将CO2还原为HCOO-的速率高达456.0 μmol·g-1·h-1,同时选择性在98%以上。机理研究表明,Co-MOF光催化剂同时具备光响应(有机配体)和催化中心(三核Co簇),能够促进电子从有机配体到金属簇的快速迁移,三核[CoIICoIICoII]簇得到电子被还原成[CoICoIICoII],进而将吸附的CO2还原成CO或者HCOO-,而1-苄基-1,4-二氢烟酰胺(BNAH)作为电子牺牲剂被空穴氧化(图3)。
Sun等[28]将具有共轭体系的分子4,4'-二苯乙烯二羧酸与氨基相结合,成功合成了一种化学稳定性优异、CO2吸附量大、可见光吸收范围广、光催化效率高的Zr-MOFs(Zr-SDCA-NH2)。氨基化以后,有机配体H2SDCA-NH2的最大吸收波长从400 nm红移至600 nm,而Zr-SDCA-NH2具有与H2SDCA-NH2配体相类似的UV-vis光谱,这说明氨基对于提高H2SDCA-NH2对可见光的响应能力起着至关重要的作用。采用Mott-Schottky测试计算可知,Zr-SDCA-NH2和H2SDCA-NH2的CB电位分别为-0.44和-0.58 V(vs NHE),均低于CO2还原成HCOO-的还原电位(-0.28 V vs NHE),而荧光猝灭现象则说明有机配体在光照下激发出的电子能够很快地转移到Zr-O簇,因此CO2的还原反应主要发生在Zr-O簇,同时在有机配体上也有可能发生(图4)。
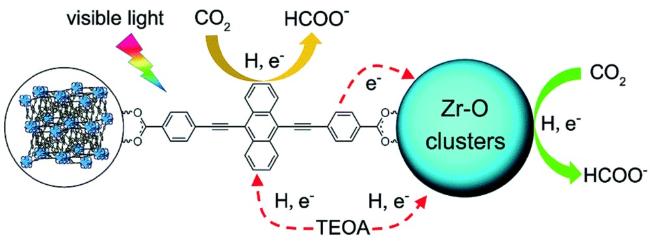
除了氨基的功能化修饰,配体中共轭体系增大也会使MOFs的吸收峰红移,从而提高其在可见光下的催化性能[33, 34]。2016年,Chen等[35]使用蒽基羧酸配体合成了一种锆基MOFs材料(NNU-28),其能够对小于650 nm波长范围的可见光表现出良好的响应能力。在可见光照射下反应10 h,以三乙醇胺作为牺牲试剂,CO2还原成HCOO-的平均速率可以达到183.3 μmol·h-1·mmo 。而且在三个测试循环后,其活性基本保持不变。通过一系列的对照试验以及ERP的测试结果表明,蒽基配体作为光响应中心,在光照下产生的光生电子可以经LMCT(linker-to-metal charge transfer)过程迁移到Zr-O簇还原CO2,同时在配体上也可以发生CO2的还原反应,而NNU-28优异的催化性能主要得益于这两种并存的催化途径(图5)。
2.2 金属配合物作为光敏剂
2.2.1 卟啉/金属卟啉
Xu等[39]合成了一种非常稳定的介孔锆基卟啉MOF(PCN-222),其具有较高的比表面积和CO2吸附量。PCN-222很好地继承了卟啉配体的可见光吸收能力,在200~800 nm波长范围内展现出较强的光吸收强度。超快瞬态吸收光谱和光致发光光谱证实PCN-222中电子陷阱态的存在可以有效抑制光生电子空穴对复合,从而显著提高CO2还原成HCOO-的光催化转化效率,在10 h的活性测试中生成HCOO- 30 μmol。根据ESR信号的变化可以推测出PCN-22光还原CO2的催化机理如图6所示,卟啉配体H2TCPP作为光吸收中心,吸光后产生的光生电子经过LMCT过程迁移到Zr-O簇,发生CO2的还原反应,产物为HCOO-,而电子给体TEOA在光生空穴处被氧化成醛。
当反应是在强酸或者强碱等比较苛刻的催化条件下进行时,催化剂的化学稳定性显得尤为重要。Chen等[40]采用自上向下的合成策略,成功制备了两个具有类似nbo拓扑结构的ZrPP-n(卟啉配体为THPP时,n=1;卟啉配体为THBPP时,n=2,)。其中,在不同浓度的HCl和NaOH溶液中浸泡一周,ZrPP-1的结构依旧完好,甚至是在饱和NaOH溶液中仍然可以稳定存在。通过后合成金属化,ZrPP-1-Co表现出最高的催化效率,这主要归因于其带隙较窄,而且Co的引入使得光生电荷的分离效率增强,有利于光催化反应的进行。
Liu等[41]成功地将Rh-卟啉均匀且规则地分散到MOFs骨架中,合成了一种具有良好热和化学稳定性的Rh卟啉MOFs(Rh-PMOF-1(Zr)),在可见光照射下能够以高达99%的选择性将CO2还原为HCOO-。在卟啉环中引入Rh(Ⅲ)以后,PMOF的激发态寿命从ns延长至μs,
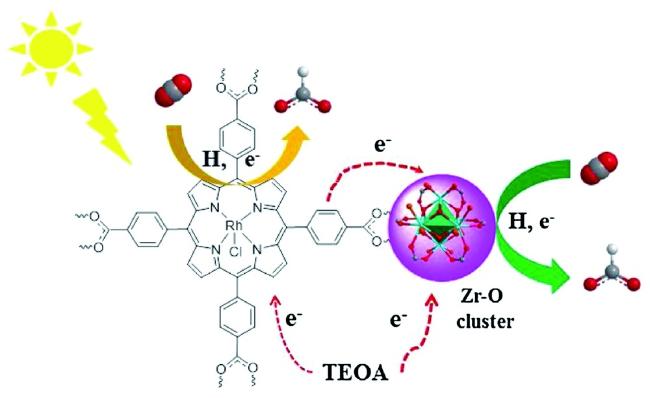
在真空下的寿命达到了207 μs,而在空气中由于单线态氧的形成,寿命减少至13 μs,这也说明Rh-PMOF-1的激发态是三重激发态。Monte Carlo模拟结果表明CO2主要吸附在卟啉环和Zr6O8簇上,通过含时密度泛函理论(TD-DFT)计算以及Mott-Schottky(MS)测试证实,TCPP-Zr6O8和Rh(TCPP)Cl-Zr6O8的前沿轨道电子主要分布在卟啉环而并非Zr6O8簇,CO2的还原反应可以在Rh卟啉环和Zr6O8簇上同时发生。如图7所示,可见光照射下,Rh卟啉配体吸光后,光生电子空穴对快速分离,光生电子转移到金属Rh以及Zr6O8簇(LMCT过程)参与CO2的还原反应,空穴被TEOA消耗。
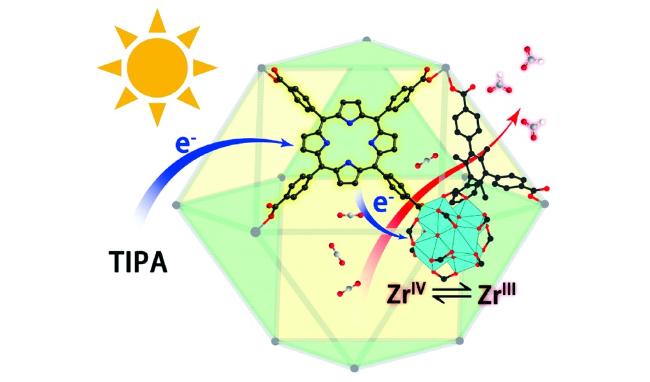
Qiu等[42]成功设计并构建了两种含有混合配体的阿基米德多面体MOFs,PCN-137和PCN-138。由卟啉配体和Zr-O簇合成的PCN-138表现出更高的光还原CO2催化性能。PCN-138的BET比表面积为1261 m2·g-1,而且在温度为273和298 K时,CO2的饱和吸附量分别为63.07和40.72 cm3·g-1,较高的吸附量有利于提高催化剂表面反应物的浓度,促进催化反应的进行。在可见光照射下,以三异丙醇胺(TIPA)作电子牺牲剂,PCN-138可以高选择性地将CO2还原成HCOO-,而配体催化的均相体系中并未发现HCOO-的存在。在催化体系中同时发现了ZrⅣ和ZrⅢ的EPR信号,证实Zr-O簇是催化反应的活性中心(图8)。
2.2.2 金属多吡啶配合物
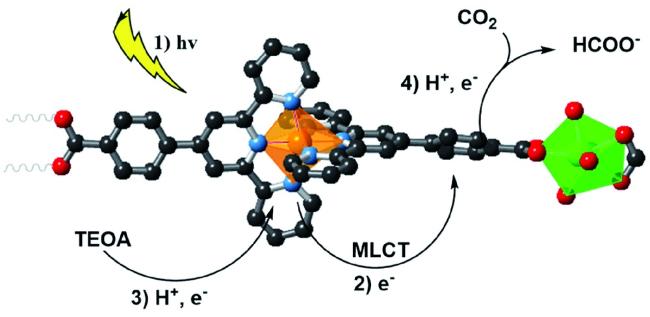
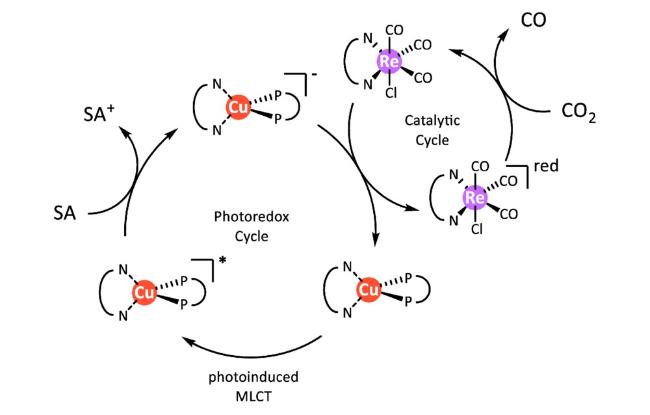
虽然多吡啶Ru和Ir配合物是非常理想的光敏剂,但是Ru和Ir地球储量有限,价格比较昂贵且毒性大。因此,基于非贵金属配合物的催化剂在实际应用中更具优势。Feng等[55]以地球上储量丰富的CuI配合物作为光敏剂,以Re配合物作为CO2还原反应的活性中心,合成了mPT-Cu/Re催化剂。MOFs在该体系中的主要作用是稳定CuI配合物以及提供一种电子从光敏剂到活性中心的有效传输途径。在350~700 nm波长的光照下反应48 h,TON值达到1328,是均相体系在同等条件下的95倍。通过一系列的光物理和电化学实验进一步研究了荧光猝灭过程,发现BIH(苯并咪唑)的存在可以有效地猝灭Me2LCu的荧光发射。mPT-Cu/Re光还原CO2的机理如图11所示,光敏剂Cu-PS在光照下变成激发态[Cu-PS]*,随后被BIH还原成[Cu-PS]-,[Cu-PS]-通过MOFs骨架将电子传递给Re活性中心将CO2还原成CO,而[Cu-PS]-失去电子重新回到基态。
3 MOFs复合材料
单一MOFs光催化剂存在很多不足,如对可见光的响应能力差和光生电子-空穴易复合。通过将MOFs与其他材料相结合,使得光生电子能够快速地在复合材料内部的迁移,从而实现高效的光生电荷分离效率,提高其催化性能。
3.1 MOFs/半导体复合催化剂
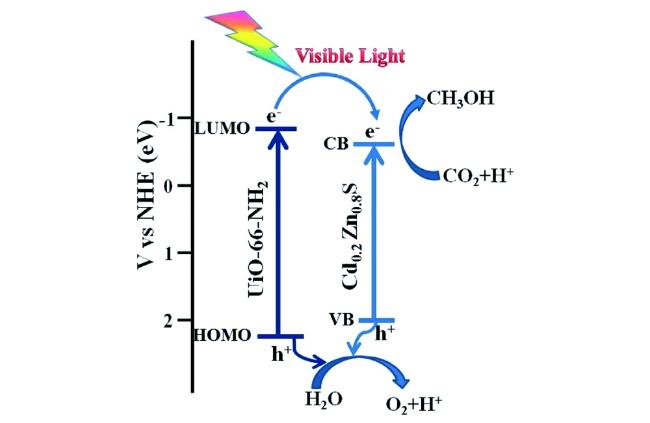
Su等[61]报道了一种新的复合物光催化剂Cd0.2Zn0.8S@UiO-66-NH2,PL和光电流测试结果表明,Cd0.2Zn0.8S和UiO-66-NH2之间异质结的形成,使得复合催化剂具有优异的光生电荷分离和迁移效率。Cd0.2Zn0.8S和UiO-66-NH2在可见光照射下均可以产生光生电子,由于UiO-66-NH2的LUMO电位(-0.60 eV vs NHE)比Cd0.2Zn0.8S的CB电位(-0.56 eV vs NHE)更负,因此光生电子可以从UiO-66-NH2的LUMO直接转移到Cd0.2Zn0.8S的CB,吸附态的CO2得到电子被还原成CH3OH,同时留在UiO-66-NH2的HOMO以及Cd0.2Zn0.8S的VB上的空穴被牺牲试剂H2O消耗,使反应能够连续进行(图12)。
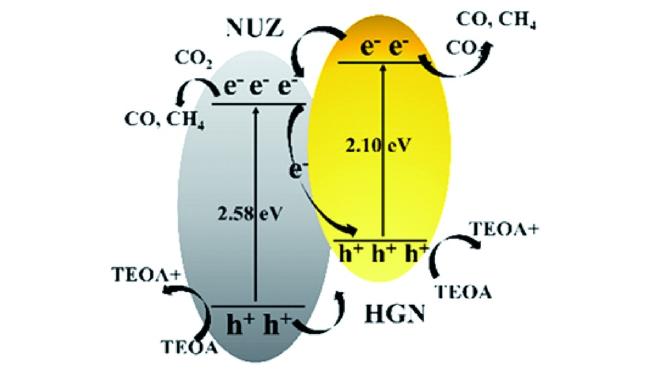
半导体与MOFs之间电子的迁移效率会在很大程度上影响其光催化活性[62, 63]。Wang等[64]通过NHx-Zr-O化学键将NH2-UiO-66固定在具有高密度氨基(—NHx)的三维多孔g-C3N4(HGN)表面(NUZ-HGN-x,x为HGN的质量分数),两者之间的强相互作用不仅能够有效抑制反应过程中NH2-UiO-66的浸出,还能够促进电子在界面处的转移。与苯环相比,HGN共轭π结构上的电子更容易发生离域,表面原位生长NUZ后,HGN和NUZ共轭π体系中的离域电子重新分配,通过影响NUZ的Zr-O簇和—NH2的电子状态增强复合催化剂的光吸收能力。同时,NUZ和HGN独特的结构表面能够多次散射和反射入射光,从而提高光的收集能力。光电流和电化学阻抗谱(EIS)测试说明,当HGN的负载量为35%时,光生电荷的分离和迁移效率最高,光催化性能最高,在可见光照射下12 h,CO的平均生成速率可以达到31.6 μmol·g-1·h-1。由Mott-Schottky测试可知HGN和NUZ的CB电势分别为-1.24和-0.65 eV vs NHE,比CO2/CO的还原电势更负,因此催化反应可以发生。如图13所示,电势差的存在使得电子可以从HGN的CB迁移到NUZ的CB,而空穴由NUZ的VB迁移到HGN的VB,可以有效抑制电子空穴对的复合。
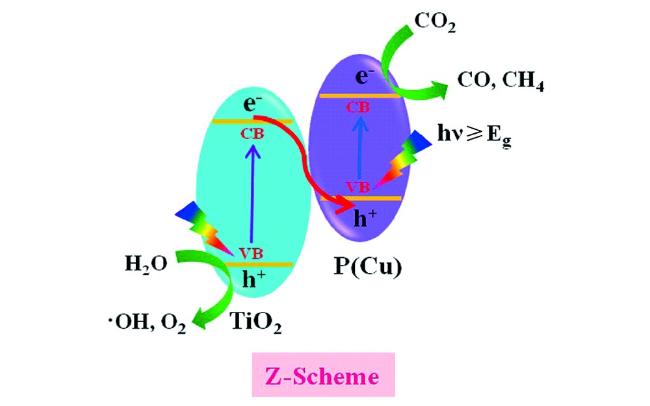
Wang等[65]采用原位溶剂热法构建了Z型PCN-224(Cu)/TiO2复合催化体系。SEM可以观察到大多数TiO2颗粒均匀地生长在MOFs外表面。当加入的TiO2的量为0.6 g时,15% P(Cu)/TiO2表现出最优的催化性能。在特定波长(λ > 300 nm)的光照下,CO的生成速率能够达到37.21 μmol·g-1·h-1,且选择性保持在99%以上。光电流测试中,复合催化剂的光电流强度分别是TiO2和PCN-224(Cu)的130和13倍,说明复合之后电荷迁移效率有了很大的提高,并通过EIS和PL的测试结果进行了佐证。以5,5-二甲基-1-吡咯啉-N-氧化物(DMPO)为自由基捕捉试剂,光照下,在TiO2和PCN-224(Cu)催化的反应体系中分别观察到了DMPO-·OH和 DMPO-·O2的ESR信号,复合催化剂催化下两种自由基的ESR信号均有出现。结合Motty-Schottky电势测试结果,他们提出了如图14所示的Z型催化机理,TiO2和PCN-224(Cu)吸光后均可产生光生电子,TiO2 CB上的电子通过接触面迁移到PCN-224(Cu)的VB,这一过程有利于提高光生电荷的分离效率,而CO2的还原反应以及H2O的氧化反应分别发生在PCN-224(Cu)的CB以及TiO2的VB。
3.2 MOFs/钙钛矿量子点复合催化剂
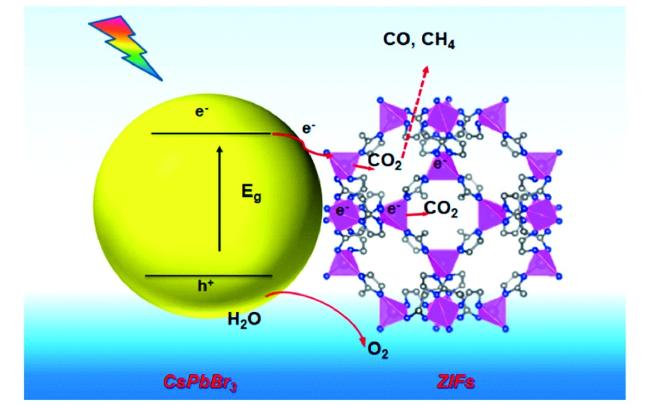
Kong等[69]采用简单的原位合成的方法在CsPbBr3量子点表面生长一层Zn或Co-ZIF材料(ZIF-8和ZIF-67),随着ZIF壳层厚度的增加,CsPbBr3量子点在水中的稳定性逐渐增强。但是过厚的ZIF壳层不仅使反应物和产物的扩散难度增加,还会影响CsPbBr3量子点对光的有效吸光,不利于光催化反应的进行。XPS、TRPL和飞秒瞬态吸收光谱(fs-TA)的结果显示,Pb2+与咪唑配体之间的强相互作用使得核与壳之间的距离缩短,从而促进光生电子在CsPbBr3和ZIF之间的快速迁移。通过估算得到的CsPbBr3@ZIF-67中电子的迁移速率比CsPbBr3@ZIF-8快一个数量级,原因在于ZIF-67的CoII在被来自配体的光生电子还原后可以作为CO2还原的催化中心,因此CsPbBr3@ZIF-67具有更高的催化活性(图15)。
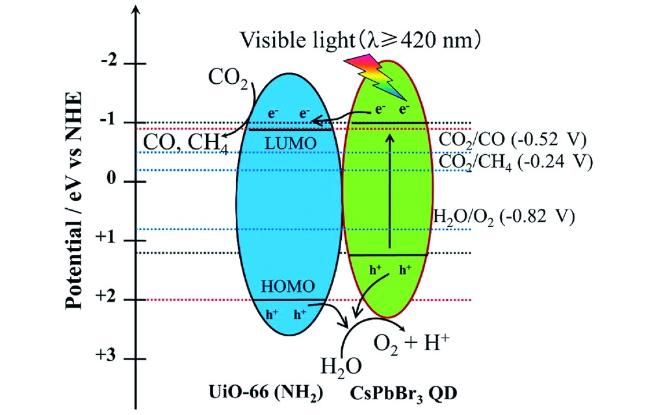
Wan等[70]采用简单的超声混合的方法合成了CsPbBr3-QDs/UiO-66(NH2)复合光催化剂,当CsPbBr3-QDs的负载量为15%时,在可见光照射下反应12 h,CO的生成速率达到8.21 μmol·g-1·h-1,而且在经历了三个反应循环后,复合催化剂仍然能够保持非常优异的催化性能。荧光猝灭和光电流强度的增强说明CsPbBr3-QDs/UiO-66(NH2)复合催化剂能够有效促进光生电荷的分离和迁移,抑制电子空穴对的复合。根据价带X射线光电子能谱(VB-XPS)和UV-vis的测试结果计算可知,CsPbBr3的CB比UiO-66(NH2)的LUMO电势更负,因此在可见光照射下,CsPbBr3 CB上的光生电子将会迁移到UiO-66(NH2)的CB上,吸附在MOFs表面的CO2被还原为CO。在空穴处发生水的氧化反应,以补充反应体系消耗的电子(图16)。
Wu等[71]将CH3NH3PbI3(MAPbI3)封装到铁卟啉MOFs(PCN-221(Fex))中,MOFs材料能够缩短Fe活性中心与钙钛矿光敏中心之间电荷转移的距离,通过TRPL证明MAPbI3/PCN-221(Fe0.2)表现出与PCN-221(Fe0.2)类似的快速荧光衰减过程,意味着光生电荷的分离和迁移速率得到增强。以水做牺牲试剂,在300 W氙灯(λ>400 nm)照射下进行了80 h的活性测试,MAPbI3@PCN-221(Fe0.2)催化下,CO和CH4的总收率高达1559 μmol·g-1,是PCN-221(Fe0.2)的38倍。而PCN-221(Fe0.2)在30 h后开始发生分解,说明MAPbI3 QD对光的竞争吸收能够减少PCN-221(Fex)的光降解失活。
3.3 MOFs/贵金属复合催化剂
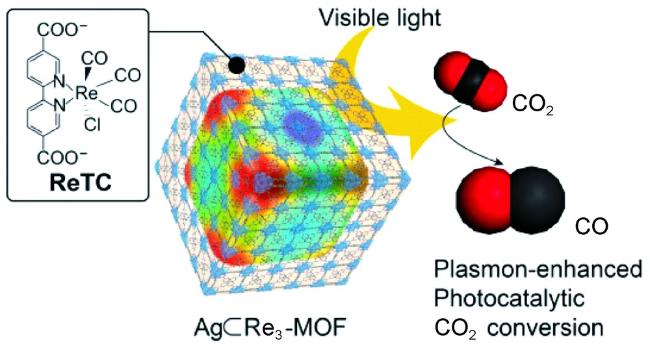
贵金属纳米粒子通过表面等离子体共振,在可见光区域表现出较强的光吸收能力,从而在表面产生高浓度的载流子,提高光催化反应的效率[72, 73]。2017年,Yaghi等[74]通过将Re3-MOF与等离子体Ag纳米颗粒结合来提高其光还原CO2的催化活性(图17)。在UV-vis光谱中可以观察到Ag纳米颗粒具有很强的局域表面等离子体共振(LSPR)散射峰(λmax~480 nm),而Ag⊂Re3-MOF复合材料依然保留有Ag纳米颗粒的LSPR特性,在可见光照射下,Ag纳米立方体表面增强的近场被Re配合物活性中心吸收,从而表现出更高的催化性能。当Ag⊂Re3-MOF复合催化剂的壳层厚度适宜(16 nm)时,其光催化活性是Re3-MOF的7倍。在连续48 h的稳定性测试中,Ag⊂Re3-MOF的催化性能始终比较稳定。相比之下,Cu纳米颗粒在可见光下不具有LSPR特性,因此Cu⊂Re3-MOF的催化活性没有明显的增强。
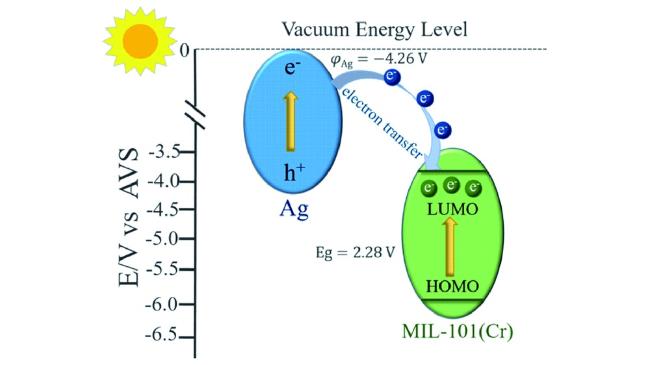
Guo等[75]通过对不同纳米尺寸(80~800 nm)的MIL-101(Cr)-Ag催化性能的研究发现,随着催化剂尺寸的减小,光还原CO2的催化性能逐渐增强。当粒径减小到80 nm时,复合催化剂表现出最高的催化活性,可见光照射下,CO和CH4的生成速率能够达到808.2和427.5 μmol·g-1·h-1,分别是粒径为800 nm催化剂的23和18倍。由扩展X射线吸收精细结构(EXAFS)光谱的分析结果可知,粒径为80 nm催化剂中Ag的配位数(N=8.95)要高于Ag NPs(N=5.71),这意味着催化剂中Ag颗粒表面的缺陷状态较少,而缺陷位处容易发生电子空穴对的复合,因此,光生电荷的分离效率显著提高。而小尺寸催化剂的边和拐角处晶胞密度更大,更有利于电子的迁移,在光电流测试中粒径为80 nm催化剂的光电流强度最大。他们通过Mott-Schottky测试进一步阐述了MIL-101(Cr)-Ag光催化还原CO2的过程,计算可知MIL-101(Cr)的LUMO和HOMO电位分别为-3.76和6.04 V(vs AVS),Ag的功函数为-4.26 V(vs AVS),如图18所示,在可见光照射下,Ag表面激发的电子将会迁移到MIL-101(Cr)的LUMO,同MIL-101(Cr)的光生电子一起还原CO2。
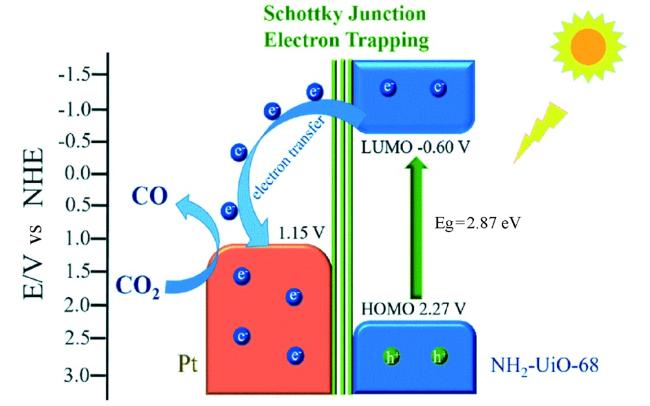
Guo等[76]设计并制备了一种具有肖特基结的核壳结构复合催化剂Pt@NH2-UiO-68,这种结合方式可以引导电子从NH2-UiO-68定向迁移到Pt,从而抑制电子空穴对的复合,提高光还原CO2的催化活性。TRPL测试表明,当Pt的含量2 wt%时,核壳结构的Pt(2)@NH2-UiO-68复合催化剂光生电荷的分离效率最高,平均荧光寿命仅有0.26 ns,可见光照射下,光催化CO2还原生成CO的速率为400.2 μmol·g-1·h-1,是Pt(2)/NH2-UiO-68催化下的3倍多。他们通过Mott-Schottky测试更加深入地研究催化还原机制,NH2-UiO-68的LUMO和HOMO电位分别是-0.60和2.27 V(vs NHE),Pt的能级为1.15 V(vs NHE),具体的电子迁移过程如图19所示,在可光照射下,肖特基结能够引导光生电子从 NH2-UiO-68的LUMO转移到金属Pt活性中心,CO2得到电子被还原成CO,空穴被TEOA消耗。
3.4 MOFs/酶复合催化剂
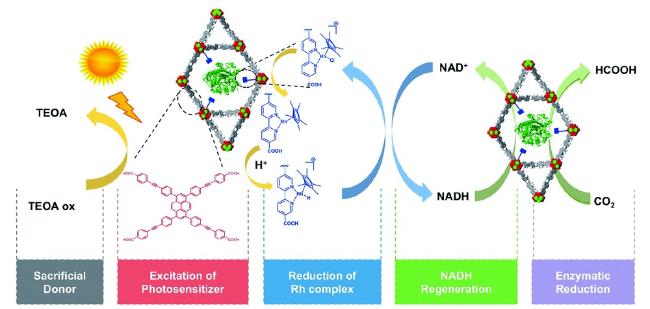
酶催化具有高选择性、高效、条件温和等优点,但是酶的活性在很大程度上受限于其特定的辅酶。因此,在酶催化还原CO2的反应中辅酶的再生起着至关重要的作用[77, 78]。2020年,Chen等[79]将NADH辅酶的还原中心Cp*Rh(bpy)Cl与甲酸脱氢酶(FDH)以及具有吸光能力的有机配体同时组装到同一MOFs框架中,制备了一种多功能复合催化剂FDH@Rh-NU-1006,可以同时实现了辅酶NADH的连续再生以及CO2的催化还原。如图20所示,在白光下照射,芘基配体激发的电子传递给电子介体,活化后的Rh基配合物将NAD+还原成NADH,还原速率约为28 mM·h-1,固载的FDH使用还原得到的辅酶NADH将CO2还原成HCOO-,周转频率(TOF)高达865 h-1。
4 MOFs衍生材料
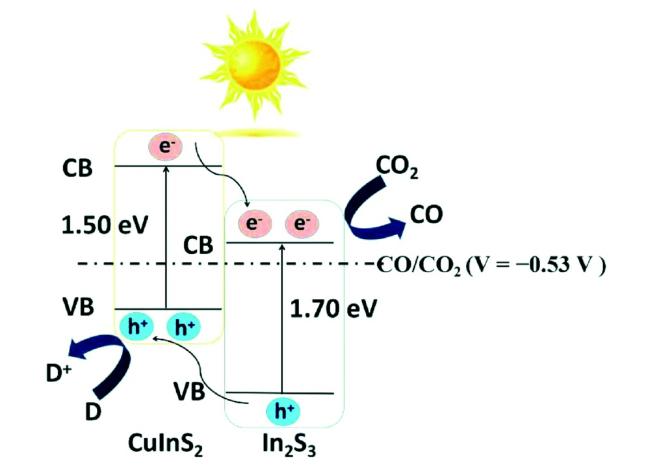
Yang等[83]以In-MOF为前驱体,通过硫化和离子交换制备了一种具有异质结的多维In2S3-CuInS2光催化剂,最优条件下,In2S3-CuInS2-15光还原CO2生成CO的速率能够达到19 μmol·h-1·g-1。PL、光电流实验以及EIS测试结果显示,由于异质结的存在,光生电子空穴对的复合受到抑制,载流子能够更快地从催化剂的内部迁移到表面参与氧化还原反应。因为CuInS2的CB(-1.535 V vs NHE)比In2S3的CB(-0.732 V vs NHE)更负,CuInS2的光生电子经界面转移到In2S3的CB,与来自In2S3 的VB上的电子一起参与CO2的还原反应(图21)。
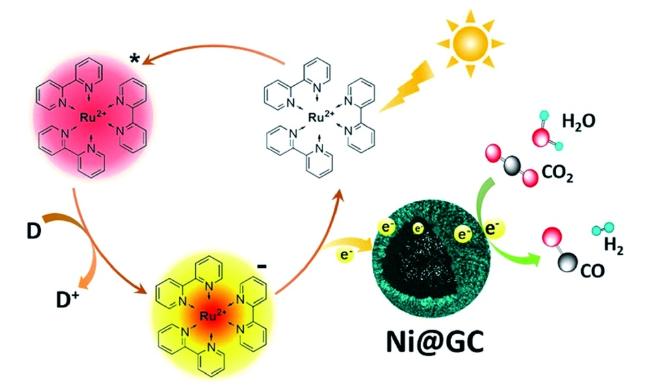
Lin等[85]以Ni-MOF为前驱体合成了空心球型Ni@GC催化剂,石墨碳壳层可以有效隔离相邻的金属Ni,防止聚集失活。结果表明在Ni和C的界面处形成的Ni—C键有利于电子从碳壳层到Ni核的迁移,富电子的Ni活性中心能够促进CO2的吸附或活化,从而表现出更高的CO2还原活性。他们通过XPS、DFT计算、紫外光电子能谱(UPS)和PL等测试手段对催化机理进行了深入研究,结果如图23所示。吸光后,激发态的[Ru(bpy)3]2+*被TEOA还原为[Ru(bpy)3]+,[Ru(bpy)3]+激发出的电子发生离域并传递给Ni@GC催化剂,Ni—C键的存在使电子富集在Ni中心,有效促进了CO2的吸附或活化,CO的生成速率为9.0 mmol·h-1·g-1,但是由于产氢副反应的存在,选择性仅有75%。而且他们发现,当把催化剂中粒径较大的Ni NPs去除以后,CO的产率会进一步提高至11.67 mmol·h-1·g-1。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
5 结论和展望
MOFs是一类新兴的多孔晶态材料,在光催化领域具有广阔的应用前景。近年来报道了很多新型MOFs基光还原CO2催化剂(单一MOFs、MOFs复合材料和MOFs衍生材料),通过对MOFs框架的功能化修饰、与其他功能材料的复合等可以显著提高其光催化性能。与传统的催化剂相比,MOFs基光催化剂具有以下优势:(1)超高的比表面积和可调的孔结构有利于CO2的吸附;(2)金属节点和有机配体易于进行针对性的功能化修饰;(3)丰富的孔道可以容纳功能客体物质;(4)MOFs衍生材料继承了母体MOFs的结构特征,同时提供更多暴露的活性位点。
但是,现阶段MOFs基光还原CO2催化剂仍然存在很多缺点和不足:(1) 可见光利用率依然很低;(2) 稳定性差,在经历数个测试循环后,催化剂的结构很难保持完整,活性下降很快;(3) 仍然需要使用有机牺牲试剂(如TEOA)、有机溶剂,这无疑会给环境带来不利影响; (4) 产物的选择性差;(5) 缺乏深入的机理研究;(6) MOFs基催化剂的制备条件重复性不佳。未来MOFs基CO2光催化的研究应重点解决上述问题,通过对MOFs材料的功能化修饰、与光敏材料的复合等提高其催化性能以及稳定性,并利用先进的原位表征技术对光催化CO2还原的反应机理进行深入研究。