




1 引言
2 冠醚的磺化方法
3 磺化冠醚对金属离子的键合
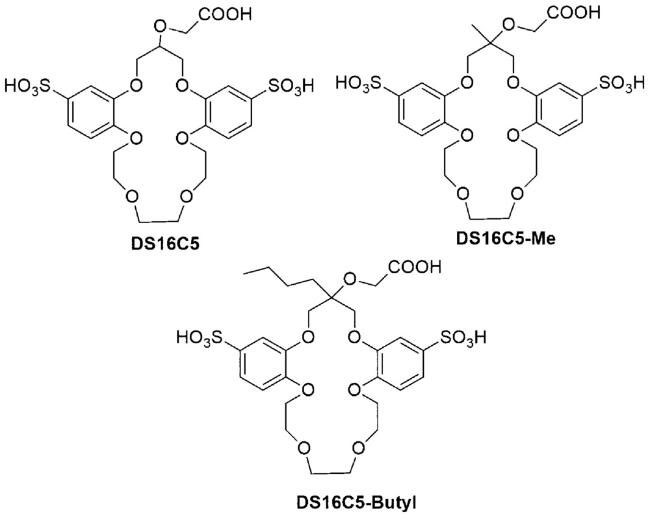
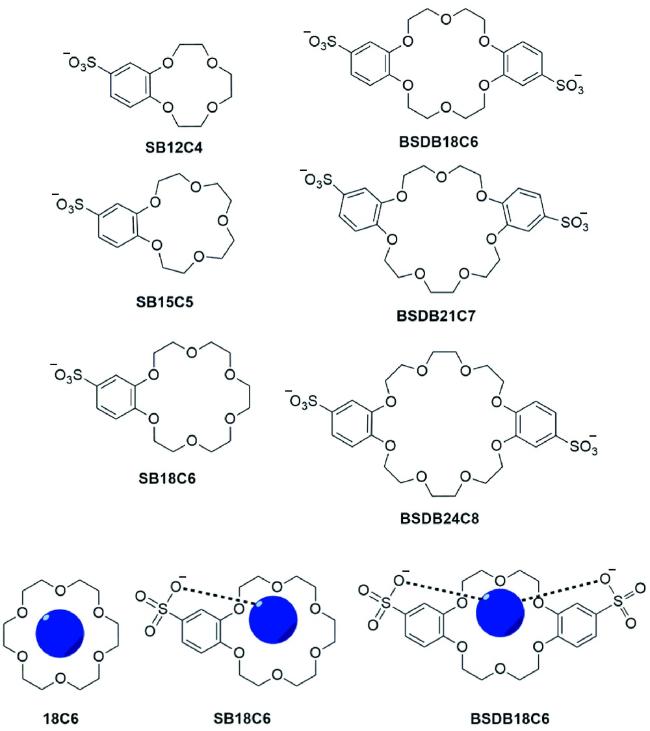
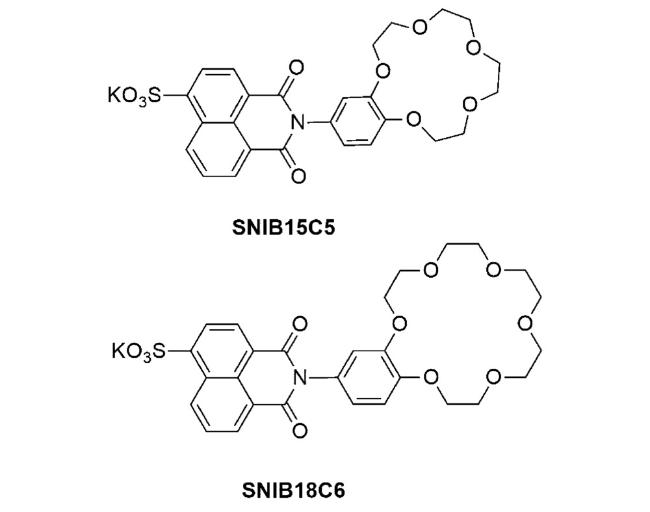
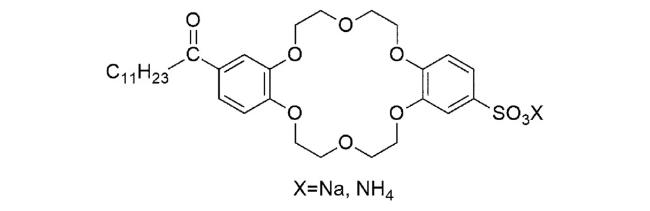
4 磺化冠醚对有机阳离子的键合与组装
4.1 二磺化冠醚对吡啶类阳离子的键合与组装
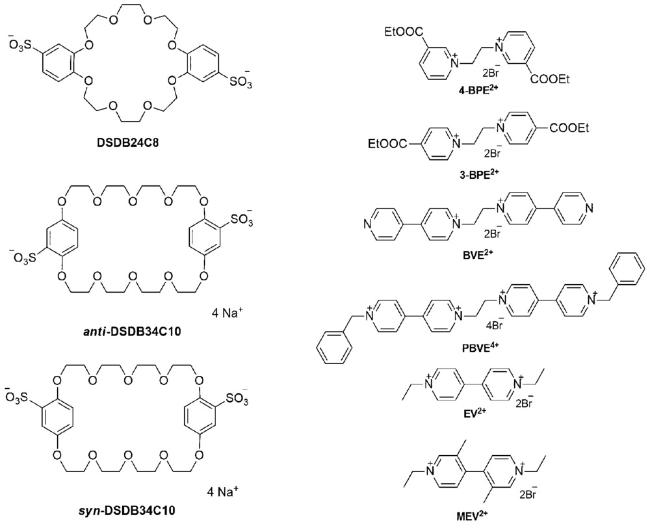
表1 在不同溶剂中形成[2]准轮烷的键合常数Ka/103 M-1[a]对比[32]Table 1 A comparison of association constants Ka/103 M-1[a] for the formation of [2]pseudorotaxanes in various solvents at 298 K[32] |
| CD3CN[b] | CD3OD[c] | CD3OD[c] | CD3OD/D2O/(CD3)2SO[d] | |
|---|---|---|---|---|
| DB24C8 | ||||
| 4-BPE2+ | 1.9 | 5.4 | [e] | 0.1 |
| 3-BPE2+ | 4.7 | 12.9 | [e] | 0.7 |
| BVE2+ | 0.9 | 2.2 | [e] | 0.1 |
| PBVE4+ | 1.0 | [e] | [e] | 0.2 |
| DSDB24C8 | ||||
| 4-BPE2+ | [e] | >100 | 0.2 | 0.5 |
| 3-BPE2+ | [e] | >100 | 0.3 | 3.7 |
| BVE2+ | [e] | >100 | 0.1 | 0.5 |
| PBVE4+ | [e] | >100 | 2.3 | 7.7 |
[a] Determined by the single-point method utilizing the equation Ka=[pseudorotaxane]/[axle]2 and using equal initial concentrations of axle and wheel(2.0×10-3 M) and the integral values of the NCH2 resonances(1H NMR spectroscopy) for complexed and uncomplexed axles. Errors are estimated to be 10% or less. [b] Values in CD3CN are for B salts. [c]Axles are employed as Br- salts. [d] A mixture of 50% CD3OD, 40% D2O. [e] One or more components are insoluble in this solvent. |
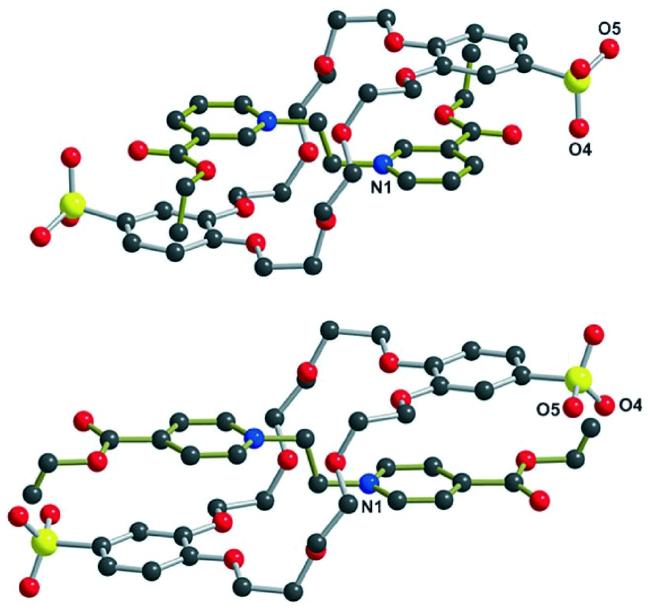
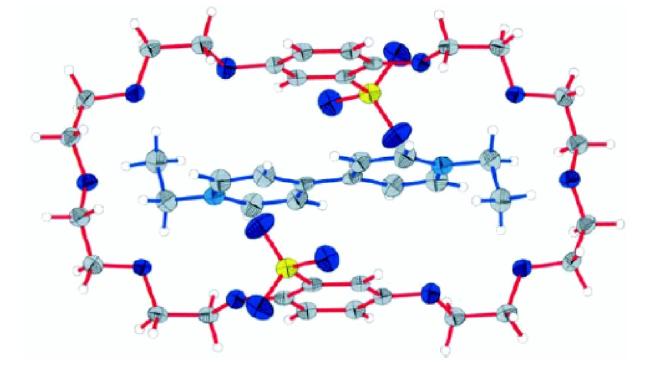
图7 4-BPE2+⊂DSDB24C8(下)和3-BPE2+⊂DSDB24C8(上)的球棍模型图。S黄色,O红色,N蓝色,轴=金色键,轮=银色键;氢原子省略[32]Fig.7 Ball-and-stick representations of the X-ray structures of [4-BPE2+⊂DSDB24C8](bottom) and [3-BPE2+⊂DSDB24C8](top). S yellow, O red, N blue, C black, axle=gold bonds, wheel=silver bonds; hydrogen atoms omitted for clarity[32] |
4.2 四磺化冠醚对阳离子的键合与组装
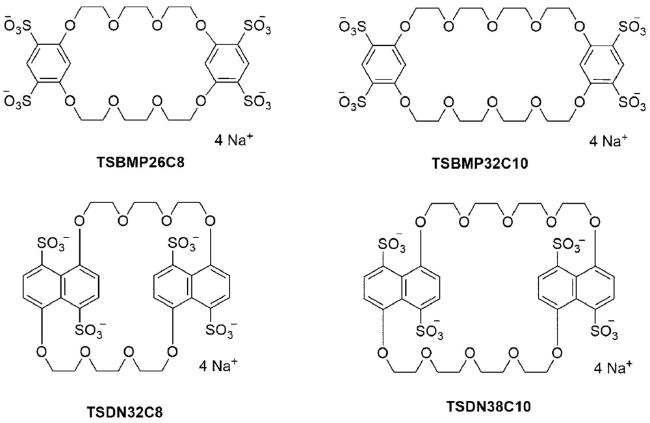
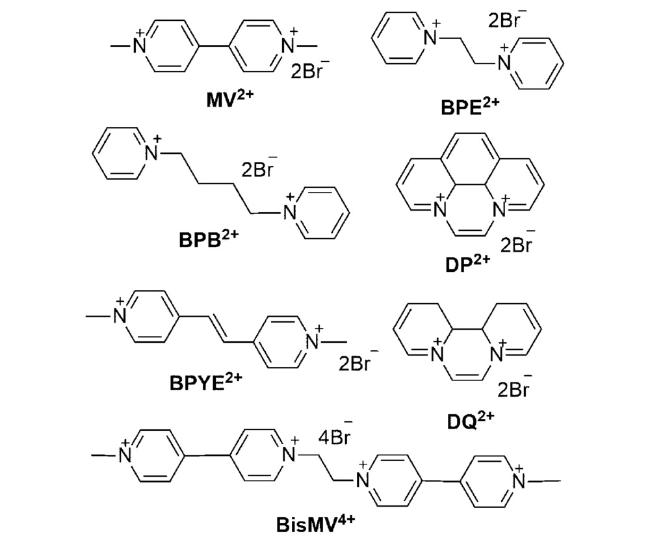
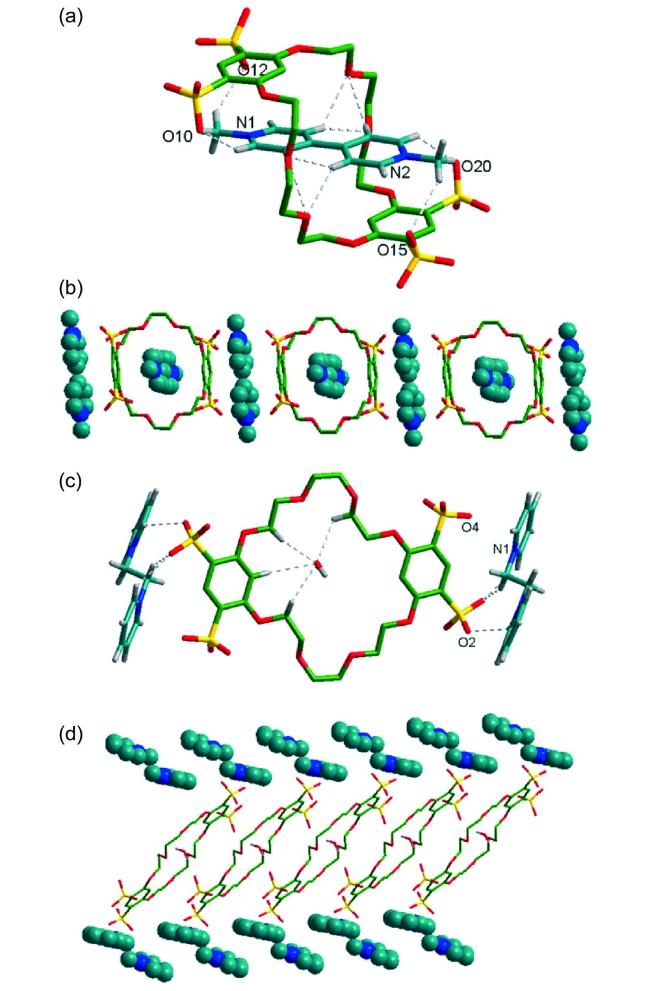
表2 TSBMP26C8 或TSBMP32C10与有机阳离子客体在水中1∶1形成络合物的键合常数(Ka/M-1)及焓(ΔH/kJ·mol-1)熵(TΔS/kJ·mol-1)变化[16, 19]Table 2 Complex associate constants(Ka/M-1), enthalpy(ΔH/kJ·mol-1) and entropy changes(TΔS/kJ·mol-1) for 1∶1 inclusion complexation of TSBMP26C8 or TSBMP32C10 with organic cationic guests in water at 25 ℃[16, 19] |
| Host | Guest | Ka(M-1) | DΔG° | ΔH°(kJ/mol) | TΔS°(kJ/mol) |
|---|---|---|---|---|---|
| TSBMP26C8 | MV2+ | (1.83 ± 0.01) × 103 | -18.62 ± 0.01 | -10.18 ± 0.08 | 7.80 ± 0.07 |
| BPE2+ | (3.44 ± 0.13) × 102 | -14.47 ± 0.09 | -3.03 ± 0.03 | 11.46 ± 0.14 | |
| BPB2+ | (2.65 ± 0.15) × 102 | -13.82 ± 0.14 | -4.26 ± 0.22 | 9.55 ± 0.36 | |
| DQ2+ | (1.63 ± 0.02) ×103 | -5.85 ± 0.03 | 12.48 ± 0.06 | ||
| DP2+ | (4.62 ± 0.08) × 103 | -10.19 ± 0.08 | 10.73 ± 0.12 | ||
| BPYE2+ | (6.52 ± 0.09) ×103 | -24.89 ± 0.05 | -3.12 ± 0.08 | ||
| BisMV2+ | (4.08 ± 0.06) ×105 | -11.53 ± 0.01 | -20.49 ± 0.19 | ||
| TSBMP32C10 | MV2+ | (4.36 ± 0.04) ×103 | -26.43 ± 0.09 | -5.66 ± 0.12 | |
| BPE2+ | (1.02 ± 0.00) ×103 | -8.02 ± 0.01 | 9.17 ± 0.00 | ||
| BPB2+ | (1.59 ± 0.13) ×103 | -11.90 ± 0.95 | 6.36 ±1.16 | ||
| DQ2+ | (3.55 ± 0.12) ×103 | -16.69 ± 0.37 | 3.57 ± 0.45 | ||
| DP2+ | (2.89 ± 0.01) ×104 | -32.58 ± 0.32 | -7.11 ± 0.31 | ||
| BPYE2+ | (1.04 ± 0.01) ×104 | -34.12 ± 0.09 | -11.19 ± 0.08 |
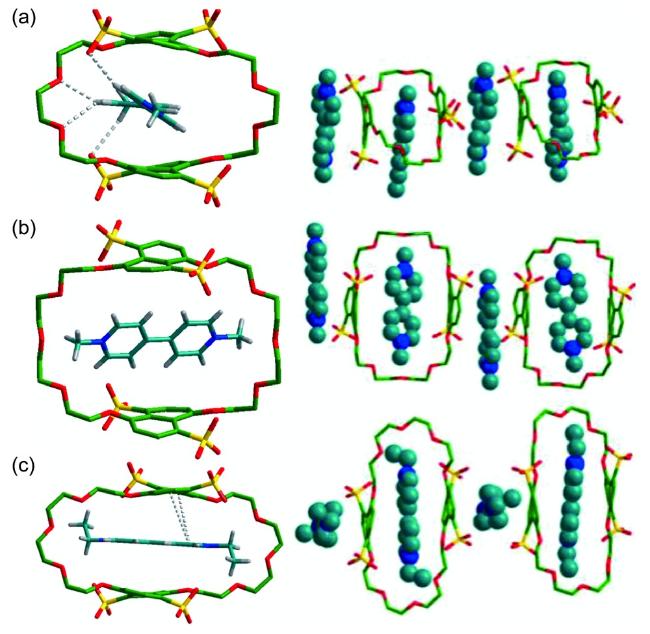
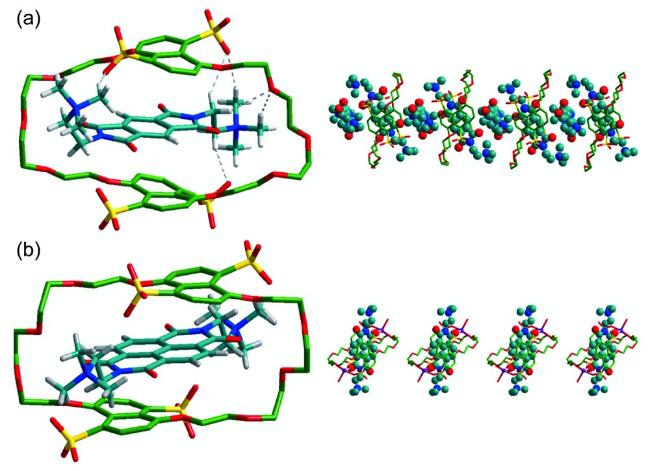
图14 [2]准轮烷(a) PMDI2+⊂TSDN38C10 和(b) NDI2+⊂TSDN38C10的晶体结构及堆积模式图,左:单晶结构,右:堆积结构。溶剂分子和部分氢原子已省略[43]Fig.14 Crystal structures and packing representation of [2]pseudorotaxane(a) PMDI2+⊂TSDN38C10 and(b) NDI2+⊂TSDN38C10. Left: single unit; right: packing structure. Solvent molecules and partial hydrogen atoms are omitted for clarity[43] |
表3 TSDN32C8 或TSDN38C10与有机阳离子客体在水中1∶1形成络合物的键合常数(Ka/M-1)及焓(ΔH/kJ·mol-1)和熵(TΔS/kJ·mol-1)变化[19, 37, 43, 44, 46]Table 3 Complex associate constants(Ka/M-1), enthalpy(ΔH/kJ·mol-1) and entropy changes(TΔS/kJ·mol-1) for 1∶1 inclusion complexation of TSDN32C8 or TSDN38C10 with organic cationic guests in Water at 25 ℃ [19, 37, 43, 44, 46] |
| Host | Guest | Ka(M-1) | D-ΔG° | - ΔH°(kJ/mol) | TΔS°(kJ/mol) |
|---|---|---|---|---|---|
| TSDN32C8 | MV2+ | (4.04 ± 0.35) × 107 | 43.40 ± 0.22 | 38.93 ± 0.27 | 4.47 ± 0.05 |
| EV2+ | (5.25 ± 0.58) × 107 | 44.04 ± 0.28 | 41.54 ± 0.54 | 2.50 ± 0.26 | |
| BuV2+ | (4.66 ± 0.48) × 107 | 43.75 ± 0.26 | 43.92 ± 1.05 | -0.17 ± 0.79 | |
| MP2+ | (1.13 ± 0.06) × 105 | 28.84 ± 0.04 | 29.23 ± 0.23 | -0.39 ± 0.35 | |
| PMDI2+ | (5.82 ± 0.05) × 105 | 32.90 ± 0.02 | 24.85 ± 0.01 | 8.05 ± 0.03 | |
| NDI2+ | (9.81 ± 0.08) × 105 | 34.18 ± 0.02 | 23.17 ± 0.03 | 11.01 ± 0.08 | |
| G1 | (2.82 ± 0.21) × 106 | 36.81 ± 0.18 | 35.47 ± 0.05 | 1.34 ± 0.23 | |
| G2 | (4.94 ± 0.29) × 106 | 38.20 ± 0.14 | 33.38 ± 0.04 | 4.82 ± 0.18 | |
| G3 | 4.32 × 106 | 37.85 | 41.39 | -3.54 | |
| G4 | (8.09 ± 0.09) × 105 | 33.72 ± 0.03 | 39.37 ± 0.37 | -5.65 ± 0.39 | |
| G5 | (1.64 ± 0.08) × 106 | 35.47± 0.12 | 40.54 ± 0.07 | -5.08 ± 0.20 | |
| G6 | (1.82 ± 0.13) × 106 | 35.52 ± 0.02 | 41.42 ± 0.42 | -5.89 ± 0.40 | |
| TSDN38C10 | MV2+ | (3.25 ± 0.04) × 105 | 31.46 ± 0.03 | 30.13 ± 0.24 | 1.33 ± 0.21 |
| EV2+ | (1.85 ± 0.04) × 105 | 30.06 ± 0.05 | 27.20 ± 0.01 | 2.86 ± 0.07 | |
| BuV2+ | (1.88 ± 0.02) × 105 | 30.10 ± 0.03 | 27.27 ± 0.01 | 2.83 ± 0.02 | |
| MP2+ | (4.42 ± 0.26) × 102 | 15.03 ± 0.69 | 14.71 ± 1.29 | 0.38 ± 1.43 | |
| PMDI2+ | (8.08 ± 0.30) × 104 | 27.99 ± 0.09 | 20.59 ± 0.15 | 7.40 ± 0.24 | |
| NDI2+ | (2.33 ± 0.03) × 106 | 36.32 ± 0.03 | 36.31 ± 0.04 | 0.01 ± 0.01 | |
| BV2+ | (7.12 ± 0.01) × 105 | 33.37 ± 0.00 | 30.99 ± 0.07 | 2.54 ± 0.07 | |
| DP2+ | (2.49 ± 0.00) × 106 | 36.47 ± 0.00 | 29.80 ± 0.06 | 6.84 ± 0.06 | |
| DMDAP2+ | (1.12 ± 0.03) × 108 | 45.89 ± 0.06 | 47.84 ± 0.12 | -1.96 ± 0.18 | |
| DBDAP2+ | (2.25 ± 0.03) × 107 | 41.93 ± 0.04 | 40.06 ± 0.06 | 1.87 ± 0.10 |
表4 五种荧光染料与四磺化二萘并-32-冠-8键合的荧光光谱的参数[19]Table 4 Parameters of fluorescence spectra of five silk tricyclic fluorescent dyes binded to tetrasulfonated dinaphtho-32-crown-8[19] |
| Dyes | Ka(M-1) | λex | I | λem | Φ | λem(complex) | Φcomplex |
|---|---|---|---|---|---|---|---|
| AO | 1.8×106 | 450 | ↑ | 527 | 0.139 | 541 | 0.186 |
| PY | 1.7×106 | 530 | ↓ | 566 | 0.303 | 572 | 0.330 |
| MB | 1.8×106 | 610 | ↓ | 693 | 0.032 | 695 | 0.033 |
| AD | 1.4×106 | 400 | Quench | 476 | 0.284 | 476 | 0.374 |
| ADZ+ | 1.0×106 | 390 | Quench | 404 | 0.429 | 404 | 0.346 |
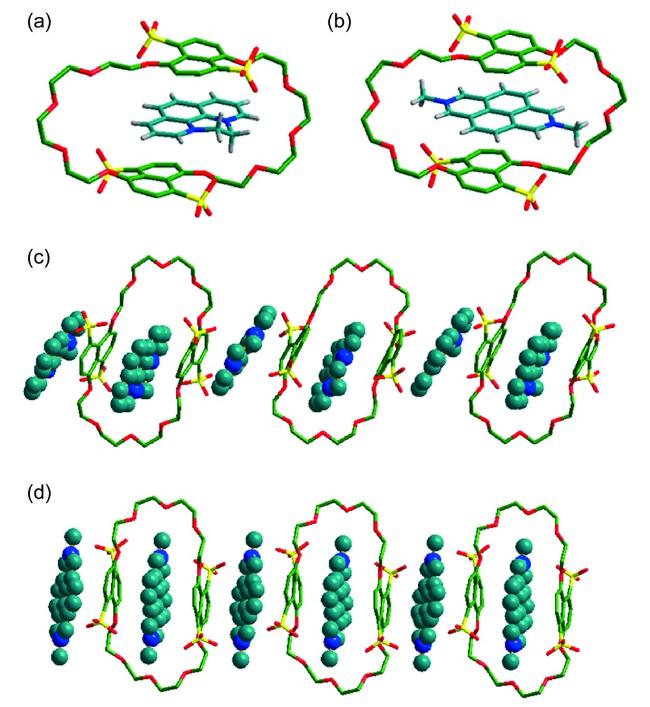
图15 (a) DP2+⊂TSDN38C10- 和(b) DMDAP2+⊂ TSDN38C10-晶体结构图;(c) DP2+⊂TSDN38C10 和(d) DMDAP2+⊂TSDN38C10的堆积模式图(溶剂分子和部分氢原子已省略)[44]Fig.15 Crystal structures of(a) DP2+⊂TSDN38C10 and(b) DMDAP2+⊂TSDN38C10, and packing representation of(c) DP2+⊂TSDN38C10 and(d) DMDAP2+⊂TSDN38C10. Please note that the solvent molecules and partial hydrogen atoms are omitted for clarity[44] |
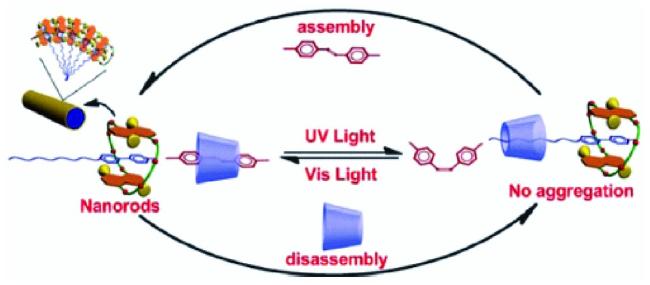
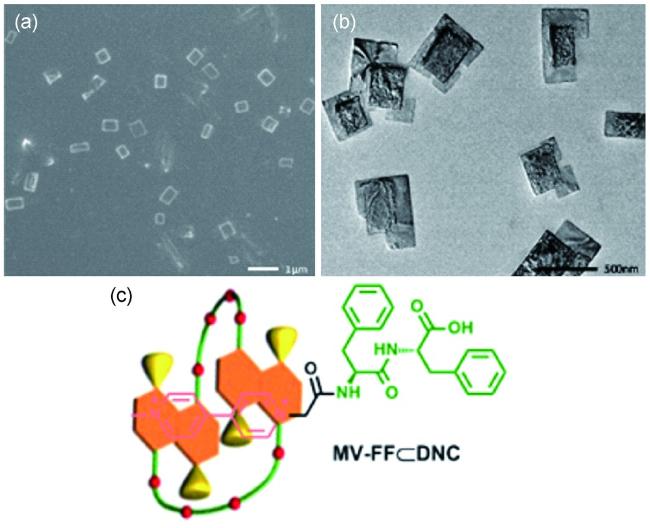
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}