




1 引言
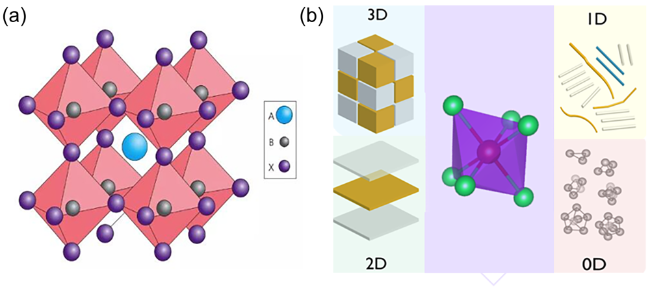
图1 (a)钙钛矿结构,(b)钙钛矿结构分类的示意图,红色球体:金属中心,绿色球体:卤化物原子[11]1978年,Weber[1]首次引入甲胺,形成了具有三维结构的有机-无机杂化钙钛矿材料。2009年时CH3NH3PbI3被用于制作染料敏化太阳能电池[2],效率为3.8%。到2012年有机-无机杂化钙钛矿实现了10.9%[3]的光电转化效率。开始引起研究人员的广泛关注。该类钙钛矿具有高吸光系数、较低的激子束缚能、长的载流子扩散长度。随后几年中,有机-无机杂化钙钛矿太阳能电池得到了快速发展,目前单节钙钛矿太阳能电池的效率已达到25.2%[4]。 Fig.1 Schematic diagram of (a) perovskite structure,(b) perovskite structure classification. red spheres: metal centers; green spheres: halide atoms[11] |
2 零维CsPbX3量子点
2.1 合成方法
2.1.1 热注射法
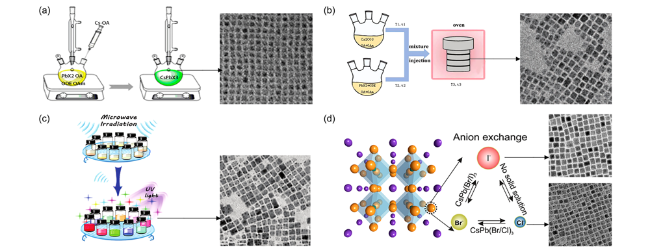
图2 (a)热注射法原理图和合成的CsPbBr3量子点TEM图,标尺100 nm[6],(b)溶剂热法原理图和合成的CsPbCl3 TEM图,标尺50 nm[21],(c) 微波法原理图和合成的CsPbBr3量子点TEM图,标尺50 nm[22],(d)离子交换原理图和合成的CsPbI3和CsPbCl3 TEM图,标尺50 nm[10]Fig.2 Schematic diagram of(a) hot injection method and TEM image of synthesized CsPbBr3 quantum dots, scale bar 100 nm[6],(b) solvothermal method and TEM image of synthesized CsPbCl3, scale bar 50 nm[21],(c) microwave method and TEM image of synthesized CsPbBr3 quantum dots, scale bar 50 nm[22],(d) anion exchange and TEM image of synthesized CsPbI3 and CsPbCl3 quantum dots, scale bar 50 nm[10] |
2.1.2 溶剂热法
2.1.3 微波法和离子交换
表1 不同条件下合成的CsPbX3量子点Table 1 CsPbX3 quantum dots synthesized under different conditions |
| Component | Temperature ( ℃) | Ligiand | atmosphere | Size(nm) | Preparation method | PL(nm) | FWHM(nm) | PLQY(%) | ref |
|---|---|---|---|---|---|---|---|---|---|
| CsPbI3 | 170 | OA, OAm TOP | Vacuo | 11.0 | Hot-injection | 679~692 | 31~39 | 100 | 26 |
| 60~185 | OA, OAm 1∶1 | N2 | 3.4~12.5 | Hot-injection | 585~670 | - | 21~55 | 29 | |
| CsPbBr3 | 170 | OA, OAm 1∶2 | Ar | 8.0 | Hot-injection | 460~510 | <30 | 60~90 | 45 |
| 140~200 | OA, OAm 1∶1 | N2 | 4.0~15.0 | Hot-injection | 410~530 | 12~42 | 50~90 | 6 | |
| 120 | OA, OAm 1∶10 | N2 | 4.0 | Hot-injection | - | 170 | 24 | 24 | |
| 190 | OA, OAm 4∶1 | N2 | 13.2 | Hot-injection | - | 103 | - | 24 | |
| - | OA, OAm 1∶1 | Air | 10.0 | Microwave | 520 | 13~37 | 10.9~92.1 | 22 | |
| Mn∶CsPbCl3 | 200 | OA, OAm 1∶1 | - | Solvothermal | 600 | 100 | 0.1~10.8 | 21 | |
| 150 | OA, OAm 1∶1 | N2 | 8.6±0.5 | Hot-injection | 600 | - | 12.7 | 35 | |
| CsPb1~xFexCl3 | OA, OAm TOP | Ar | 6.7~7.8 | Hot-injection | 401~403 | 13.8~14.6 | 6.2 | 40 | |
| CsPbBr2I | 90 | OA, OAm TOP | Air | 7.7 | Solvothermal | 632 | 14~43 | - | 20 |
| CsPbBrI2 | 90 | OA, OAm TOP | Air | 7.7 | Solvothermal | 632 | 14~43 | - | 20 |
| CsPb(Br/I)3 | 40 | OA, OAm 1∶1 | - | 4.0~15.0 | Ion exchange | 508~688 | 12~40 | 10~80 | 10 |
| CsPb(Br/Cl)3 | 40 | OA, OAm 1∶1 | - | 4.0~15.0 | Ion exchange | 404~508 | 12~40 | 10~80 | 10 |
2.2 粒径控制
2.2.1 工艺温度的影响
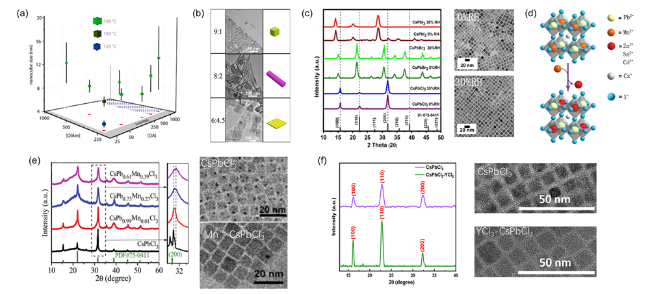
图3 (a) 不同油酸油胺配比及不同温度下合成的CsPbBr3粒径[24],(b)不同油酸:油胺比例下制备的CsPbBr3纳米晶[27],(c) 不同湿度下合成的CsPbX3 XRD图和相应的CsPbBr3 TEM图[32],(d) B位元素掺杂示意图[35],(e) Mn2+掺杂前后的CsPbCl3 XRD和TEM图[21],(f)使用YCl3前后的 CsPbCl3 XRD和TEM图[10]Fig.3 (a) Sizes of CsPbBr3 nanocube synthesized using various concentrations of oleylamine(OlAm) and oleic acid(OA) and different reaction temperatures[24],(b) TEM images of CsPbBr3 obtained with different ratios of OA and OAm[27],(c) XRD patterns and TEM images of CsPbX3 synthesized under different humidity[32],(d) schematic diagram of doping[35],(e) XRD patterns and TEM images of CsPbCl3 before and after Mn2+ doping[21],(f) XRD patterns and TEM images of CsPbCl3 prepared with or without YCl3[10] |
2.2.2 前驱液成分的影响
2.2.3 添加剂的影响
2.3 光学性质
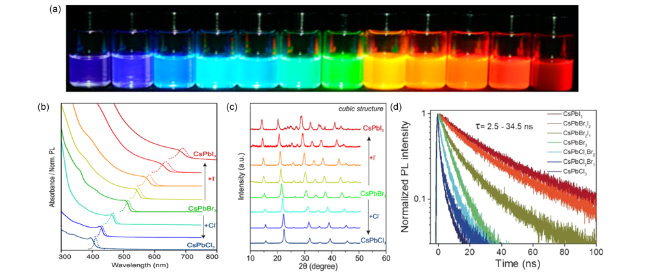
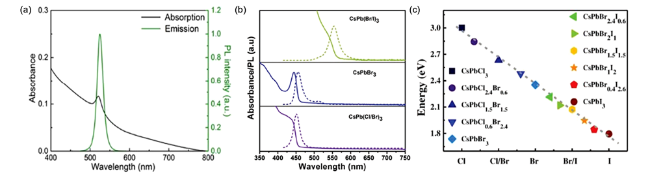
图4 (a)紫外灯(λ= 365 nm)下不同卤素成分CsPbX3在甲苯中的胶体溶液[6],(b)随着PbCl2或PbI2的增加,CsPbBr3的紫外光吸收(实线)和PL(虚线)光谱的变化[10],(c)阴离子交换各产物XRD图[10],(d)不同卤素时间分辨光致发光衰减曲线[43]Fig.4 (a) Colloidal solutions in toluene under UV lamp(λ = 365 nm)[6],(b) evolution of the optical absorption(solid lines) and PL(dashed lines) spectra of CsPbBr3 NCs with increasing quantities of PbCl2 or Pb ,(c) powder X-ray diffraction(XRD) patterns of the parent CsPbBr3 NCs and anion-exchanged samples[10],(d) time-resolved PL decay curves obtained for CsPbX3 NCs with halogen ions varying from Cl- to |
2.4 太阳能电池中的应用
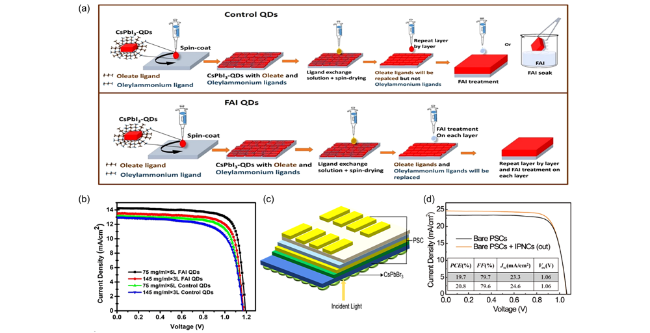
图5 (a)固态配体交换(Control QDs和 FAI QDs)示意图[46],(b)基于control QDs和FAI QDs的器件J-V曲线[46],(c)使用CsPbBr3@SiO2制备的具有外部涂层的器件结构图[50],(d)裸露和具有CsPbBr3@SiO2外部涂层的器件J-V曲线[50]Fig.5 (a) schematic diagram of solid-state ligand exchange(control QDs and FAI QDs)[46],(b) J-V curves of the control and FAI QD cells[46],(c) device structure of the PSCs coated with CsPbBr3@Si ,(d)J-V characteristics of the bare device and CsPbBr3@SiO2-coated device[50] |
表2 不同无机钙钛矿材料制备的太阳能电池性能参数Table 2 Performance parameters of solar cells made of different inorganic perovskite materials |
| Perovskite | Structure | Modification Method | Jsc (mA/cm2) | FF (%) | Voc (V) | PCE (%) | Time(h)/Attenuation % | ref |
|---|---|---|---|---|---|---|---|---|
| CsPbI3 QDs | FTO/TiO2/CsPbI3QDs/spiro-OMeTAD/Al | Pb(OAc)2 | 13.47 | 65.00 | 1.23 | 10.77 | 1440/0 | 29 |
| FTO/TiO2/CsPbI2Br/CsPbI3QDs/PTAA/Au | Mn2+ doping Pb(OAc)2 | 15.25 | 78.70 | 1.20 | 14.4 | 480/5 | 12 | |
| FTO/TiO2/CsPbI3QDs/Spiro-OMeTAD/Au | MeOAc | 14.80 | 74.00 | 1.11 | 12.15 | 1440/15 | 28 | |
| FTO/NiO/CsPbI3QDs/C60/ZnO/Ag | FAI | 14.25 | 77.60 | 1.19 | 13.10 | 40/20 | 46 | |
| CsPbBr3 QDs | FTO/ZnONPs/CsPbBr3-CsPb2Br5/Spiro-OMeTAD/Au | NH4SCN | 6.17 | 77.20 | 1.43 | 6.81 | 2400/0 | 48 |
| FTO/GQDs/CsPbBr3/CsPbBrI2QDs/carbon | - | 5.08 | 66.70 | 1.21 | 4.10 | 480/50 | 50 | |
| FTO/TiO2/CsPbBr3QDs/CH3NH3PbI3QDs/Spiro-OMeTAD /Au | - | 23.31 | 68.90 | 1.02 | 16.4 | 100/18 | 51 | |
| CsbBr3/FTO/CeTiO2/perovskite/Spiro-OMeTAD/Au/Al2O3 | - | 23.05 | 78.11 | 1.11 | 20.02 | 4000/10 | 52 | |
| FTO/TiO2/CsPbBr3QD/PTB7/Ag | GaSCN | 2.30 | 67.60 | 1.65 | 2.57 | 100/10 | 18 |
3 一维CsPbX3纳米线/纳米棒
3.1 合成方法
3.1.1 热注射法
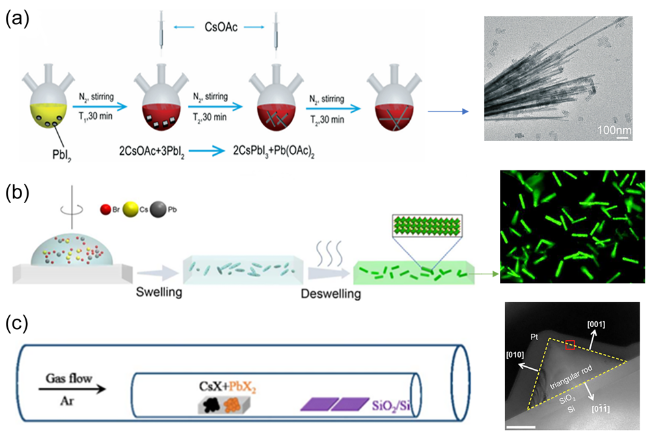
图6 (a)两次注入前驱体的热注射法原理图和相应的CsPbI3 TEM图[55],(b)SDM合成CsPbX3原理图和相应CsPbBr3纳米棒的荧光显微图,标尺20 μm [57],(c)气相沉积合成纳米线原理图和相应的CsPbBr3 TEM图,标尺500 nm[61]Fig.6 (a) Schematic diagram of hot injection method with two injections of precursor and corresponding TEM images of CsPbI3[55],(b) scheme of CsPbBr3 NRs-PM formation process through SDM(swelling-deswelling microencapsulation) and corresponding fluorescence microscopy images of CsPbBr3 nanorods, scale bar 20 μm [57],(c) schematic diagram of nanowire synthesis by vapor deposition and corresponding TEM images of CsPbBr3, scale bar 500 nm[61] |
3.1.2 溶液法
3.1.3 气相沉积法
3.2 形貌控制
3.2.1 工艺温度的影响
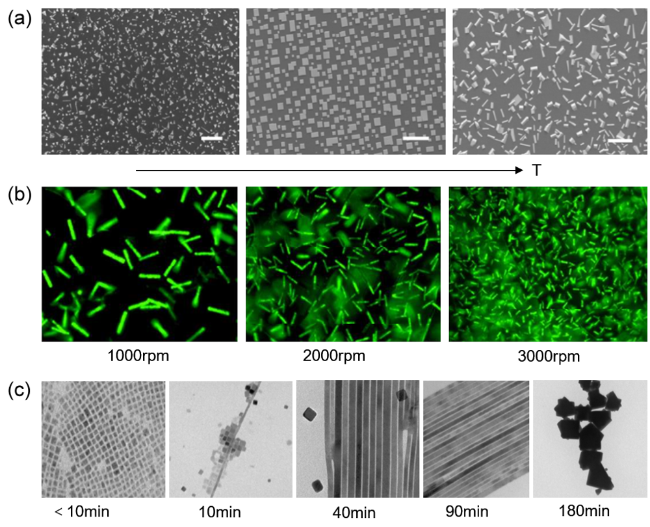
图7 (a)不同温度下制备的钙钛矿CsPbBr3纳米线SEM图,标尺20 μm [61],(b)不同搅拌速度制备的CsPbBr3纳米棒荧光显微图,标尺20 μm [57],(c)不同反应时间CsPbBr3形貌变化的TEM图,标尺100 nm[53]Fig.7 (a) SEM images of CsPbBr3 perovskite prepared with the growing temperature decreasing gradually from left to right, scale bar 20 μm [61],(b) fluorescence micrographs of CsPbBr3 prepared at different stirring speeds, scale bar 20 μm [57],(c) TEM images of morphology changes of CsPbBr3 at different reaction times, scale bar 100 nm[53] |
3.2.2 前驱液成分的影响
3.2.3 添加剂和其他因素的影响
3.3 光学性质
3.4 太阳能电池中的应用
表3 不同实验条件下合成的CsPbX3纳米线参数Table 3 The parameters of CsPbX3 nanowire synthesized under different experimental conditions |
| Component | Preparation method | Temperature ( ℃) | Ligand | Atmosphere | Length (μm) | PL (nm) | FWMH(nm) | PLQY (%) | PCE (%) | Time/% | ref |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CsPbI3 NW | Hot-injection | 250 | OA∶OAm 1∶1 | N2 | 5.00 | 446 | - | - | 53 | ||
| Two step Hot-injection | 60,120 | OA∶OAm 1∶1 | N2 | 10~20 | 685 | - | - | 55 | |||
| Solvothermal | 150 | - | Air | >1.00 | 450 | - | 0.11 | 5500/1 | 13 | ||
| CsPbBr3 NW | Hot-injection | 150 | OA∶OAm 1∶1 | N2 | 5.00 | 521 | - | - | 53 | ||
| Hot-injection | 160 | OA∶OAm 1∶1 | Ar | - | 465 | 0.15 ev | 30 | 54 | |||
| Solvothermal | 100 | OA∶OAm 1∶1 | Air | 10.00 | 520 | 14 | 75 | 15 | |||
| SDM | 25,40~60 | - | N2 | 1.27~7.15 | 526 | 18 | 23-30 | 57 | |||
| Vapor deposition | 350~380 | - | Ar | 10.00 | 530 | 0.4 | 80 | 60 | |||
| Vapor deposition | - | - | Ar | 2.00~20.00 | 534 | 22 | - | 61 | |||
| Vapor deposition | 290~330 | - | Ar/H2 | 0.15 | 539 | - | - | 63 | |||
| Ion exchange | 150 | - | >1.00 | 538 | 40 | - | 1.21 | 5500 h/1 | 13 | ||
| CsPbCl3 NW | Hot-injection | 150 | OA ∶OAm 1∶1 | N2 | 5.00 | 521 | - | - | 53 | ||
| Solvothermal | 100 | OA OAm TOP | Air | 10.00 | 410 | 16 | 12 | 14 |
4 二维CsPbX3纳米片
4.1 合成方法
4.1.1 热注射法
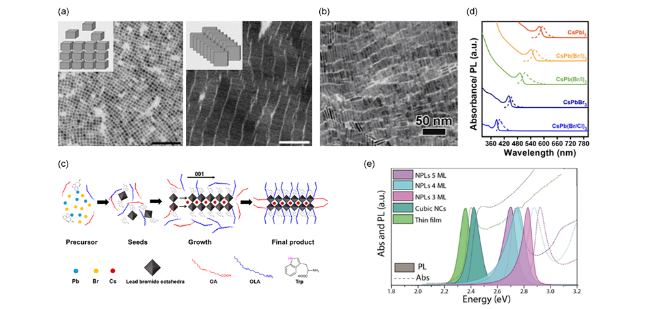
图9 (a) 不同CuCl2:PbBr2比例下合成的钙钛矿,标尺100 nm[74],(b)80 ℃下微波法合成纳米片[75],(c)色氨酸合成CsPbBr3原理图[78],(d)CsPbX3的紫外吸收光谱和PL光谱[75],(e)不同厚度CsPbBr3纳米片、立方相纳米晶、薄膜的吸收光谱和PL光谱[71]Fig.9 (a) TEM images of the perovskite nanocrystals synthesized at different CuCl2∶PbBr2 ratio, Scale bar 100 nm[74],(b) TEM images of CsPbBr3 nanoplate synthesized under 80 ℃, Scale bar 50 nm[75],(c) schematic illustration of the proposed growth mechanism for CsPbBr3 nanoplate[78],(d) UV-vis absorption(solid line) and PL emission spectra(dash line) of colloidal CsPbX3 nanoplate[75],(e) absorption and PL spectra of CsPbBr3 as a thin film, cube-shaped nanocrystals, and nanoplate of different thicknesses[71] |
4.1.2 微波法及其他
4.2 厚度控制
4.2.1 工艺温度的影响
4.2.2 前驱液成分的影响
4.2.3 添加剂及其他因素的影响
4.3 光学性质
表4 不同条件下合成纳米片的参数Table 4 The parameters of nanosheet synthesis under different conditions |
| Component | Preparation method | Temperature (℃) | Ligand | Atmosphere | Thickness (nm) | PL (nm) | FWMH (nm) | PLQY (%) | ref |
|---|---|---|---|---|---|---|---|---|---|
| CsPbBr3 | Hot injection | 90~130 | OA ;OAm 1∶1 | Vacuo | 3.0 | 470 | - | 84 | 70 |
| Hot injection | 170 | OA∶OAm 2∶1 | N2 | 3.5 | - | - | 61 | 82 | |
| Hot injection | 150 | OA∶OAm 1∶2 | N2 | 2 | 460 | - | 6~10 | 88 | |
| Solution method | 25 | OA OAm 1∶1 | Air | 1.8~3.0 | - | - | 31~78 | 71 | |
| Microwave | 160 | OA OAm 1∶1 | Air | 3.3 | 495 | 17 | 75 | 82 | |
| Solution method | 25 | OA OAm Amino acid | Air | 2.2~5.2 | 467~514 | - | 33~94 | 78 | |
| Vapor deposition | 570~600 | - | Ar | 100.0~500.0 | 510 | 22 | - | 94 | |
| CsPb(Br/I)3 | Microwave | 80 | OA OAm 1∶1 | Air | 3.2 | 520 | - | - | 75 |
| CsPb (Cl/Br)3 | Microwave | 80 | OA OAm 1∶1 | Air | 3.6 | 490 | - | - | 75 |
| CsPbI3 | Ion exchange | 25 | - | Air | 1.8~3.0 | - | - | - | 71 |
| Two-step method | 160 | OA OAm 1∶1 | - | 3.0~6.0 | 665 | - | 12 | 79 | |
| Hot injection | 100 | OA OAm 2∶5 | N2 | - | 590 | 19 | 23 | 88 | |
| CsPbCl3 | Ion exchange | 25 | - | Air | 1.8~3.0 | - | - | - | 71 |
| Recrystallization | 25 | OA OAm 1∶1 | Air | 2.2 | 355 | - | 20 | 73 | |
| Two-step method | 160 | OA∶OAm 1∶1 | - | 3.0~6.0 | 392 | 9 | 1.8 | 79 | |
| Hot injection | 80 | OA∶OAm 1∶2 | N2 | - | 384 | 12 | 0.3 | 88 |
4.4 太阳能电池中的应用
5 三维CsPbX3钙钛矿
5.1 合成方法
5.1.1 溶液法
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
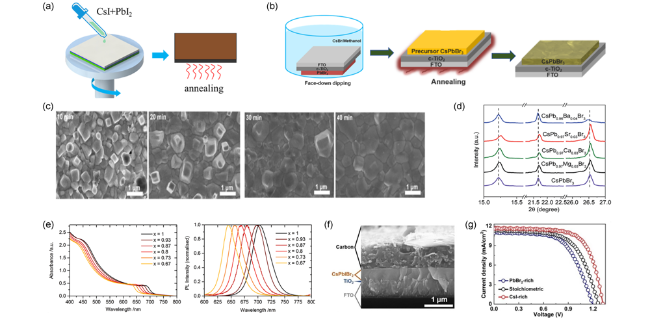
图10 (a)一步法原理图,(b)面向下液浸法原理图[98],(c)不同液浸时间制备的CsPbBr3钙钛矿TEM图[98],(d)二价路易斯酸掺杂钙钛矿XRD图[112],(e)CsPbIxBr1-x钙钛矿紫外吸收光谱、PL谱[113],(f)基于CsPbIBr2的器件截面SEM图[117],(g)不同CsI/PbBr2比例器件J-V曲线[117]Fig.10 (a) Schematic illustration of one-step method,(b) schematic illustration of the face-down dipping method[98],(c) TEM diagram of CsPbBr3 perovskite prepared by different dipping time[98],(d) XRD patterns for divalent cation doped perovskite[112],(e) UV-Vis absorption spectra and PL emission spectra of CsPbIxBr1-x[113],(f) cross-sectional SEM image of CsPbIBr2 PSC[117],(g) PCEs of CsPbIBr2 PSCs prepared with various CsI/PbBr2 stoichiometric ratios[117] |