



1 引言
自1972年Fujishima和Honda使用金红石型TiO2阳极与铂(Pt)阴极进行光电催化水分解的研究以来,研究人员为构建有效的光电催化水分解体系做出了巨大努力[7,8]。海水是地球上最丰富的自然资源,太阳能驱动海水分解制取H2是解决能源危机的理想方法[9,10]。开发光电催化海水分解过程[11],不仅能使海水资源得到有效利用,解决淡水资源匮乏的问题,还能够极大降低太阳能转换过程的成本,更加符合实际应用需求。与光电催化纯水相比,光电催化海水的优势有三点:(1)海水的储量丰富,来源广泛,有助于缓解能源危机;(2)海水中含有较多无机盐成分,在光电催化海水体系中能够提高电解液的电导率,加快电荷转移,提高光电催化效率;(3)海水中的无机盐成分可能会对光电极工作产生促进作用。
2 光电催化海水分解制氢
2.1 光电催化基本原理
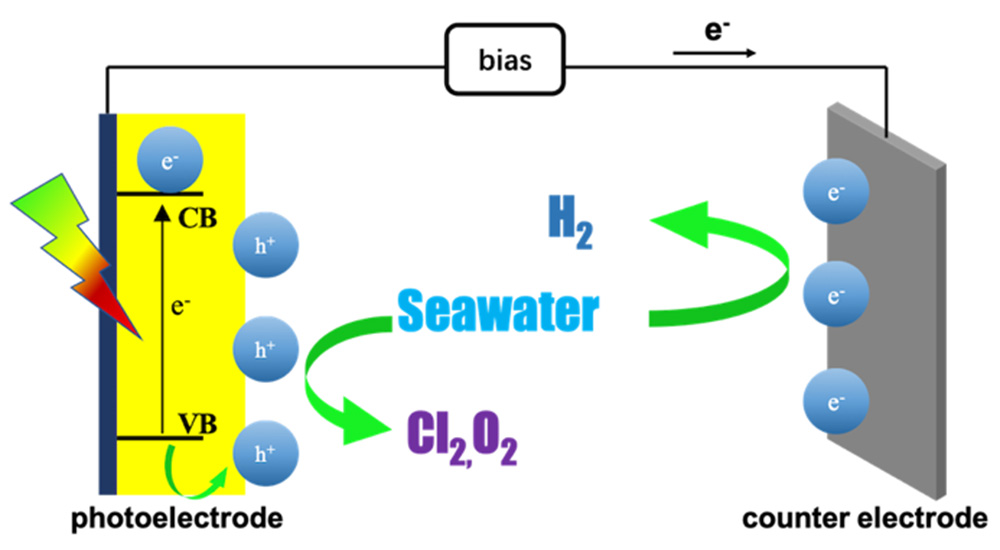
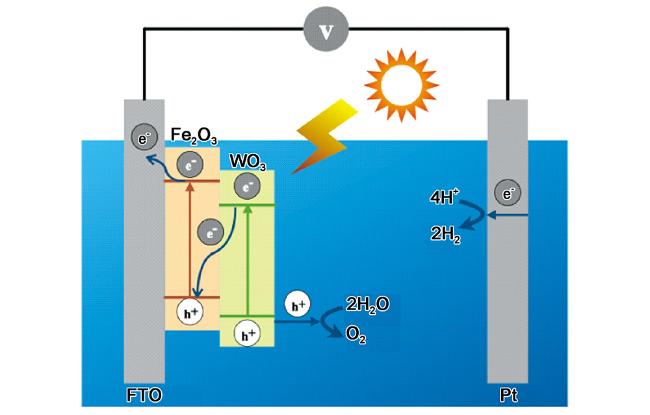
光电催化制氢是在光照下通过电解水生产H2的技术。光催化剂薄膜沉积在合适的基底上形成光阳极或光阴极,在光照下,光电极产生电子-空穴对,通过外电路流向对电极参与电极与电解质界面处的氧化、还原反应,该过程主要以电流密度和产氢速率来评价光电催化性能[12,13]。光生电流密度是衡量光电催化性能的常见表现形式,析氢速率能够表现析氢反应中的产氢性能,因此在光电催化海水分解制氢的系统中,两个性能表现都可以运用,目前大多数研究只采用其中一个,从参考文献的数据来看,Kawde等[14]报道的光生电流密度为20 mA·cm-2,析氢速率为27.5 μmol·cm-2·h-1,Yuan等[16]报道的光生电流密度为0.13 mA·cm-2,而析氢速率为30.06 μmol·cm-2·h-1。光生电流密度和析氢速率之间不存在简单的倍数关系,两者之间的联系随着光电催化系统的变化而变化,这和各种实验条件的变化密切相关。光电催化水解的工作原理如图1所示。
1997年Ichikawa[11]以透明薄膜锐钛矿TiO2作光阳极催化剂,铂作光阴极,在太阳光下,光电催化分解天然海水的析氢速率约为1.6 μmol·cm-2·h-1,光生电流密度约为0.0947 mA·cm-2。这是首篇报道光电催化分解天然海水产生H2的文章,并未施加外置电压,相应的光生电流密度偏低。该文开创了阳光与海水结合利用的先河,自此,光电催化分解海水制氢逐渐为人关注。
2.2 TiO2系材料
表1总结了TiO2体系光电催化分解海水的条件和性能。
表1 TiO2体系光电催化分解海水文献总结[10,11,14~23]Table 1 Literature summary of photoelectrocatalytic seawater splitting in TiO2 system[10,11,14~23] |
| Photoanode | Photocathode | Electrolyte | Bias | Light source (intensity, wavelength) | Photogenerated current density (mA·cm-2) | Hydrogen evolution rate (μmol·cm-2·h-1) | ref |
|---|---|---|---|---|---|---|---|
| TiO2@g-C3N4 nanorod arrays | / | natural seawater | 1.23 V vs RHE | AM 1.5 G | 1.64 | / | 10 |
| anatase TiO2 | / | natural seawater | 0 | 36.2 mW·cm-2 sunlight | 0.0947 | 1.6 | 11 |
| / | p-Si/TiO2/NiO x | simulated seawater | -0.9 V vs RHE | AM 1.5 G | 20 | 27.5 | 14 |
| / | Pt/TiO2 | concentrated seawater | solar cell | (75±5.0) mW·cm-2 UV light | / | 277 | 15 |
| PANI/GO/TiO2 ternary hybrid films | / | simulated seawater | 0.6 V vs Ag/AgCl | visible light | 0.13 | 30.06 | 16 |
| In2S3/Anatase/Rutile TiO2 dual-staggered-heterojunction nanodendrite array | / | simulated seawater | 1.23 V vs RHE | AM 1.5 G | 1.57 | / | 17 |
| TiO2 nanotube | / | concentrated seawater | 2.0 V | (74±3.4) mW·cm-2 UV light | / | 105 | 18 |
| TiO2 nanotube | / | concentrated seawater | 3.0 V | (75±5.0) mW·cm-2 UV light | / | 270 | 19 |
| Fe2O3/TiO2 | / | simulated seawater | 1.23 V vs RHE | AM 1.5 G | 0.4 | / | 20 |
| TiO2 nanotube | / | NaCl solution | 1.0 V vs Ag/AgCl | AM 1.5 G | 2.6 | / | 21 |
| TiO2 nanorod arrays | / | NaCl solution | 0.5 V vs SEC | AM 1.5 G | 2.3 | / | 22 |
| TiO2 nanotube | / | natural seawater | 3.0 V | (75±5.0) mW·cm-2 UV light | / | 215 | 23 |
TiO2由于具有适宜的能带结构、化学性质稳定、无毒和成本低等优点,受到研究人员青睐。表1中TiO2系的光催化剂主要作为光阳极材料使用,除使用各种形貌的TiO2,研究还加入Pt、Fe2O3和g-C3N4等其他材料对TiO2进行修饰形成复合材料。电极材料的性能主要表现在光生电流密度和析氢速率两个方面。
2.2.1 光阳极
光阳极催化剂产生光生电子-空穴对,空穴在光阳极参与氧化反应,电子通过外电路流向对电极参与还原反应。
TiO2的导带电位(-0.2 V vs SHE,pH=0)略高于H+的还原电位(0 V vs SHE,pH=0),不易发生还原反应,而价带电位(+3.0 V vs SHE,pH=0)远低于H2O的氧化电位(+1.23 V vs SHE),有利于发生氧化反应。因此,存在偏压时,TiO2适宜在光电催化体系中作光阳极催化剂。相比于光阴极,TiO2作为光阳极催化剂时很少使用助剂来提高性能,多为TiO2纳米管和TiO2纳米棒以及其他化合物修饰TiO2形成的复合材料。
2.2.2 光阴极
当TiO2用作光阴极时,光阴极催化剂产生光生电子-空穴对,光生电子直接在光阴极发生H+还原反应,而光生空穴通过外电路流向光阳极。
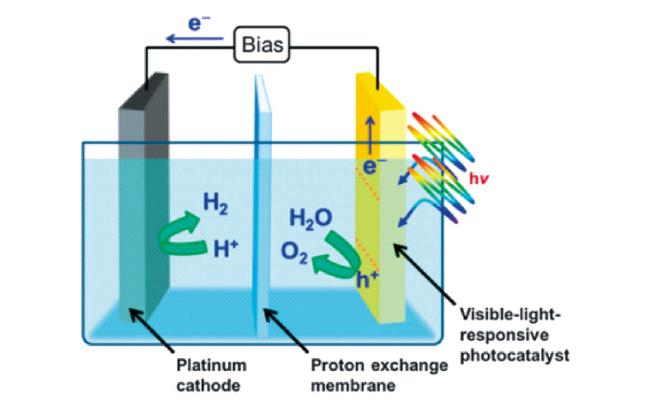
2018年Kawde等[14]报道在p-Si晶片上覆盖TiO2层,并以NiO x 作助剂得到的材料p-Si/TiO2/NiO x 作光阴极。AM 1.5 G(air mass 1.5,天顶角≈48.2°得到的太阳光谱,光强为100 mW·cm-2)下,-0.9 V vs RHE偏压,光电催化模拟海水的光电流密度为20 mA·cm-2,析氢速率约为27.5 μmol·cm-2·h-1。Nam等[15]在2011年发表的文章中,负载0.2 wt% Pt的TiO2(Pt/TiO2)作光阴极催化剂,TiO2作光阳极催化剂,在紫外光下,太阳能电池提供偏压,光电催化天然海水析氢速率为205 μmol·cm-2·h-1;光电催化浓缩海水(天然海水纳米过滤和反渗透混合滤液)的析氢速率为277 μmol·cm-2·h-1。因为电解质浓度增加,加速电极室中的电荷运动,从而溶液的电导率增加,光生电流密度和催化析氢速率随之升高。
2.2.3 电解质
表1中大多数文章使用天然海水或模拟海水作电解质,仅有少数文章使用NaCl溶液作电解质。其中Yuan等[16]使用模拟海水为0.24 M NaCl、0.055 M MgCl2和0.029 M Na2SO4的混合溶液,Yang等[17]使用的模拟海水为红海海盐溶液,虽然NaCl是海水中含量最多的溶质成分,但是海水中其他成分如MgCl2和Na2SO4等对水解过程的影响也不容忽视,如光电催化反应中生成的Mg(OH)2会附着在光阴极上阻碍反应发生。其他条件相同时,NaCl溶液的光电催化性能会优于天然海水和模拟海水,作为对海水分解机理的初步研究,可以选择该体系。从实际需求角度,对天然海水和模拟海水的研究会更真实地展现催化剂光电催化海水分解的性能。
2009年Joo等[18]报道使用海水滤液(浓缩海水)作电解液,管状氧化TiO2作光阳极催化剂,氢化酶固定的管状TiO2作光阴极,紫外光下,2.0 V偏压,光电催化过滤液、反渗透滤液和天然海水的析氢速率分别为105 μmol·cm-2·h-1、100 μmol·cm-2·h-1和81.5 μmol·cm-2·h-1。2011年,Oh等[19]使用相对紧密和宽松的纳米过滤膜NF90过滤海水得到的滤液和海水作电解质。在2 MPa压力下,NF90滤液的总溶解固体(total dissolved solids, TDS)数值高于3 MPa下NF90滤液的TDS。氧化的管状TiO2做光阳极催化剂,Pt作光阴极,波长为350~450 nm范围光照下,3.0 V偏压,光电催化2 MPa和3 MPa NF90滤液和海水的析氢速率分别为270 μmol·cm-2·h-1、240 μmol·cm-2·h-1和207 μmol·cm-2·h-1。上述结果说明,电解质浓度增加,光电催化析氢速率会随之增加,这表明相比一般海水,总溶解固体数值较高的浓缩海水是更有效的光电催化制氢的电解质。
2.2.4 偏压
2.2.5 光源
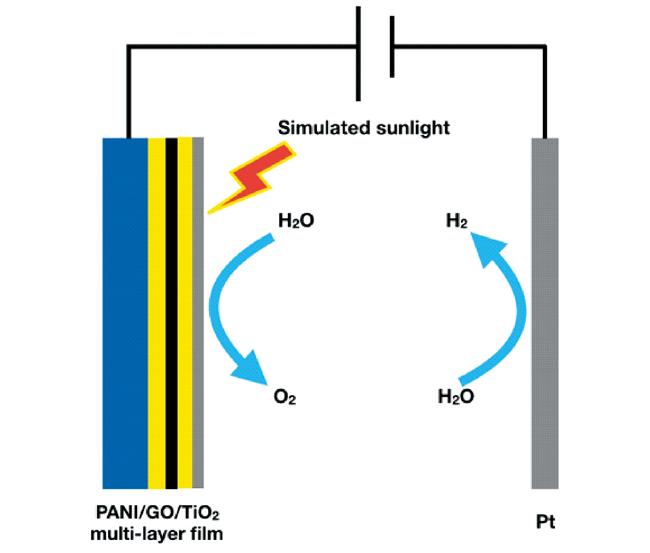
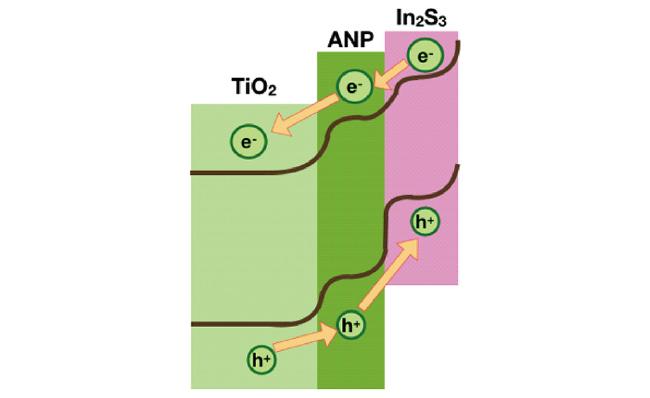
在TiO2系材料中,对太阳光或模拟太阳光的利用居多,更符合实际使用需要。2015年Li等[10]报道TiO2@g-C3N4纳米棒作光阳极催化剂,在模拟太阳光下光电催化天然海水(pH=6.4)的光电流密度为1.64 mA·cm-2。2015年Barreca等[20]报道Fe2O3沉积TiO2作阳极,复合后的TiO2能够增大对光的吸收范围,不仅降低催化剂的电荷复合率,还能够弥补TiO2对太阳光吸收效果不足的缺点,AM 1.5 G照射下,1.23 V vs RHE偏压,光电催化模拟海水的光生电流密度为0.4 mA·cm-2。Yuan等[16]在ITO电极上合成了聚苯胺(Polyaniline,PANI)-氧化石墨烯(Graphene oxide,GO)-TiO2三元混合薄膜PANI/GO/TiO2如图2所示,TiO2-GO和PANI之间的异质结使光阳极上的电子-空穴有效转移,并且增大了TiO2对光的吸收波长范围,使薄膜具有可见光响应活性。0.6 V vs Ag/AgCl偏压,光电催化模拟海水的光生电流密度为0.13 mA·cm-2,析氢速率约为72.5 μmol·cm-2·h-1。
光强越大,光源提供的光子越多,会有更多的光生电子和光生空穴产生,光催化效率在一定程度上也会增加,但是过高的光强不利于光生电子-空穴对的迁移,增加复合概率,也增大了能源的消耗,所以建议选择合适光强的光源。同时,为符合太阳能利用的实际情况,建议考察自然光下的光电催化海水性能。
2.2.6 性能
本文从电流密度和析氢速率两个角度进行了性能分析。
(1)电流密度
目前,最高的电流密度20 mA·cm-2出自Kawde等[14]的文章。由于是光阴极催化剂,光生电子直接在光阴极参与还原反应,同时促进光生电子-空穴对的分离和转移,降低电子-空穴的复合率,所以空穴能够有效地从光阴极流向对电极,他们又在催化剂上辅以助剂,最终在AM 1.5 G下获得了较高的光生电流密度。
电流密度排在第二位的是Raja等[21]在2006年报道的TiO2纳米管。他们用TiO2纳米管簇作光阳极催化剂,Pt作光阴极,AM 1.5 G的照射下,1.0 V vs Ag/AgCl偏压,光电催化NaCl溶液,产生的电流密度为2.6 mA·cm-2。文章中1.0 V vs Ag/AgCl偏压换算成可逆氢电极电势约为1.68 V vs RHE,高于水分解基本的电压,并且使用NaCl溶液而无其他离子对催化剂影响,所以光生电流密度也会相对偏高。
(2)析氢速率
Nam等[15]在2011年发表的文章中记载了最高的析氢速率。他们利用TiO2作光阴极催化剂,使用高TDS的浓缩海水作电解质,在紫外光下得到最高的析氢速率277 μmol·cm-2·h-1。因为电解质浓度增加,加速电极室中的电荷运动,从而溶液的电导率增加,并且辅以贵金属Pt作助剂,光电催化浓缩海水析氢速率随之提升。Pt/TiO2作光阴极催化剂,光生电子产于光阴极并在此发生H+还原反应,降低从外电路传递的损失,也有助于提升析氢速率。
2011年Oh等[19]利用TiO2纳米管作光阳极,紫外光下得到了光电催化浓缩海水的析氢速率为270 μmol·cm-2·h-1。同样使用浓缩海水作电解质,增加了电解质的浓度和电导率,能够使电荷快速传递,并且施加了相对高的偏压3 V,增大了电子传递驱动力,析氢速率也相应提升。
2010年Nam等[23]报道使用TiO2纳米管作光阳极催化剂,Pt为对电极,可见光下光电催化天然海水析氢速率为215 μmol·cm-2·h-1。长直状的TiO2纳米管具有良好结晶度,3.0 V偏压提供了更大的电子传递驱动力,有助于光生电子的传输,从而提升光电催化海水析氢速率。
Joo等[18]使用管状TiO2作光阳极光电催化浓缩海水的析氢速率为105 μmol·cm-2·h-1。由于电解质为浓缩海水,有利于电荷快速传输,对电极为负载氢化酶的TiO2,有利于H+在电极和电解质界面还原,光电催化海水的析氢速率有所提升。
2.3 其他材料
表2总结的是其他光电催化材料及其工作条件和性能。
表2 其他光电催化材料文献总结[6,25~32]Table 2 Literature summary of photoelectrocatalytic seawater splitting in other materials systems[6,25~32] |
| Photoanode | Photocathode | Electrolyte | Bias | Light source (intensity, wavelength) | Photogenerated current density (mA·cm-2) | Hydrogen evolution rate (μmol·cm-2·h-1) | ref |
|---|---|---|---|---|---|---|---|
| orthorhombic Ag8SnS6 | / | 0.5 M NaCl solution | 1.23 V vs RHE | AM 1.5 G | 2.5 | / | 6 |
| / | porous Co3O4 film | natural seawater | -0.96 V vs RHE | AM 1.5 G | 20 | / | 25 |
| / | ReS2 nanosheet | saturated NaCl solution | -0.25 V vs RHE | AM 1.5 G | / | 216 | 26 |
| AlB2 | / | 0.7 M NaCl solution | 0.5 V vs SHE | AM 1.5 G | 1 | / | 27 |
| WO3/g-C3N4 nanosheet arrays | / | natural seawater | 1.23 V vs RHE | AM 1.5 G | 0.73 | / | 28 |
| α-Fe2O3/WO3 nanorod arrays | / | natural seawater | 1.23 V vs RHE | AM 1.5 G | 1 | / | 29 |
| AlGaN/GaN heteroepitaxial films | / | anode: NaCl solution,cathode: natural seawater | 1.0 V vs Ag/AgCl | 300 W Xe lamp | / | 95 | 30 |
| Mo/BiVO4 | / | natural seawater | 1.0 V vs RHE | AM 1.5 G | 2.16 | / | 31 |
| BiVO4 -CoLa(OH) x | / | natural seawater | 1.0 V vs RHE | AM 1.5 G | 0.6 | / | 32 |
2.3.1 光阳极
光电催化反应器中,大部分研究的重点为光电极,对光电极的研究集中于光阳极上的光催化剂。光电催化海水体系中,与光电催化纯水相似,n型半导体在一般情况下作光阳极催化剂,吸收光子后产生电子-空穴对,电子通过外电路流向对电极并在对电极与电解质界面发生还原反应,剩余空穴在光阳极参与氧化反应。在外加偏压的作用下,加速了光生电子和空穴的分离,降低复合速率。其他材料体系中,大多数文章介绍了光阳极催化剂材料,多数为复合材料和非整比化合物,如WO3/g-C3N4和Ag7.4SnS5.3。由于海水中富含Cl-,所以在采用非TiO2材料作光阳极的光催化剂时,应该了解材料的价带与Cl-的氧化电位的位置关系,并及时检测光阳极生成的氧化产物,要善于利用Cl-在光阳极处作为牺牲剂的作用,从而提高光电催化分解海水的性能。
2.3.2 光阴极
2017年Patel等[25]报道多孔薄膜Co3O4作光阴极催化剂,能够吸收可见光,在AM 1.5 G下,外加-0.96 V vs RHE偏压,光电催化天然海水产生20 mA·cm-2光生电流密度。2019年Zhou等[26]将金属相(T)和半导体相(Td)ReS2复合形成异质结纳米片,做光阴极催化剂,在半导体相分布着Re空位(VRe),与S空位(VS)相比,由于VRe对H+的吸附能低于对其他5种金属阳离子,所以VRe比VS吸附更容易吸附H+,作为光阴极催化剂,这能够促进还原反应的进行,提高析氢速率。VRe含量6.1%的T@Td-ReS2具有最好的光电催化性能,在AM 1.5 G下,饱和NaCl溶液作电解质,施加-0.25 V vs RHE偏压,产氢速率为216 μmol·cm-2·h-1。
2.3.3 电解质
2.3.4 偏压
2.3.5 光源
只有Lee等[30]使用了300 W Xe灯作为光源,未说明光的强度和波长,其余均使用AM 1.5 G作光源。其他材料催化剂与TiO2相比,由于带隙变窄或复合材料的异质结作用,其他材料催化剂对光的吸收范围扩大,更适宜使用太阳光做光源。
2.3.6 性能
(1)电流密度
2017年Patel等[25]报道的多孔薄膜Co3O4产生最高的电流密度为:20 mA·cm-2,由于作光阴极催化剂,并且材料薄而多孔,能够降低电荷传递电阻,有利于电荷快速分离,产生高电流密度。
2016年Cheng等[6]报道采用斜方晶系的Ag8SnS6作光阳极催化剂,电流密度排在第二位。在金属前驱体中[Ag]/[Ag+Sn]比值为0.91的样品Ag7.4SnS5.3由于晶粒尺寸和晶界数量适宜,具有较低的电子-空穴复合率,所以光电催化性能较好。AM 1.5 G下,施加1.23 V vs RHE偏压,0.5 M NaCl溶液(pH=7)作电解质,光电催化产生的电流密度为2.5 mA·cm-2。
2011年Luo等[31]报道Mo掺杂并由RhO2作助剂的多孔BiVO4作光阳极催化剂,电流密度排在第三位。与单独BiVO4相比,由于Mo的掺杂增强了电导率,并且增加了孔扩散长度,而RhO2加快了光生空穴的传输速度,因此RhO2/Mo/BiVO4能够显著提高光生电流密度。在AM 1.5 G下,外加1.0 V vs RHE电压,光电催化天然海水的光生电流密度为2.16 mA·cm-2。
Li等[29]和Kravets等[27]报道的文章中电流密度均为1 mA·cm-2。2017年Kravets等报道了关于二元合金超导体(MgB2、AlB2、NbB2和NbSe2),这些材料具有电导率高、表面积大、结构灵活等优点,其中光电催化效率最高的是AlB2作阳极,使用0.7 M NaCl溶液作电解质的体系。2016年Li等[28]合成了WO3/g-C3N4纳米片簇作光阳极催化剂,电流密度排在第六位。在AM 1.5 G下,1.23 V vs RHE偏压,光电催化天然海水的光生电流密度为0.73 mA·cm-2。由于g-C3N4具有共轭大π键结构,并且存在异质结,所以能够提高电子空穴分离效率。图6展示了WO3/g-C3N4纳米片簇光阳极的工作图,光生电子从g-C3N4的导带迁移到WO3的导带,进一步通过外电路迁移到Pt电极参与H+还原反应。WO3/g-C3N4纳米片簇之间的异质结能够有效促进光生电子-空穴对的分离。

Ayyub等[32]采用BiVO4-CoLa(OH) x 作光阳极催化剂,在AM 1.5 G下,1.0 V vs RHE偏压,光电催化天然海水(pH=6.8)产生0.6 mA·cm-2光生电流密度。
(2)析氢速率
最高析氢速率出自Zhou等[26]的文章,216 μmol·cm-2·h-1。他们使用具有Re空位的金属相和半导体相的ReS2作光阴极,Re空位对H+的吸附能低于对其他五种金属阳离子,所以能够加快H+还原速率,并且两相之间存在异质结,能够加快光生电子和空穴的分离,降低复合速率。其后,Lee在2019年发表的文章[30],采用具有异质结的Al0.2Ga0.8N/GaN复合材料作光阳极催化剂,光电催化海水产氢和还原CO2生成甲酸(HCOOH)。相比于单独的GaN材料,AlGaN/GaN界面处的“二维电子气(2DEG)”能够促进光生电子从光阳极到欧姆接触电极的传输。光阴极为Sn,在装有海水的阴极室中持续鼓入CO2。阳极室电解液为1 M NaCl溶液,1.0 V vs Ag/AgCl偏压,光电催化产氢速率为95.5 μmol·cm-2·h-1。
2.4 光电催化海水机理认识
光电催化水解具有光催化和电催化两方面的特性。以光阳极为例,电极光催化剂吸收光子激发出电子-空穴对后,施加一定的偏压,光生电子会通过外电路迁移至对电极,因此抑制光生电子和空穴复合,光生空穴和电子分别在光阳极和对电极参与氧化和还原反应,当外加偏压高于水的氧化电势时,水会直接进行电化学氧化反应,这时电氧化和光催化反应会协同发生。另外,单纯地从电催化的角度来看,相比于纯水,高TDS的海水促进电极-电解质界面的离子快速传输,可提高电解质的电导率,因此光电催化分解海水制氢的速率会高于纯水。
光电催化海水制氢可以看作是在光电催化纯水的基础上,在电解质中引入了一些离子化合物,加入的离子则会对光电催化反应产生不同的影响。目前检索的文章中,涉及到对光电催化反应影响的离子主要有氯离子(Cl-)和镁离子(Mg2+)。
(1)Cl-的影响

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Luo[31]认为Cl-在光阳极会被氧化生成氯气,如公式(1);而生成的氯气易溶于水生成HClO,如公式(2);HClO会在光照下分解生成O2,如公式(3);最终光阳极检测到的O2并非来自水的氧化,而是来自上述(1)~(3)反应,这里的Cl-充当了牺牲剂的作用。光电催化海水分解主要研究光电催化析氢反应,所以光阳极发生的氧化反应不是重点,且Cl-氧化对阴极发生的H+还原不会造成负面影响。相反,空穴(h+)积极地与Cl-反应能够加快光生电子-空穴的分离,进而提升析氢速率和光生电流密度。
2Cl-+2h+→Cl2
Cl2+H2O→H++Cl-+HClO
HClO→H++Cl-+1/2O2↑
(2)Mg2+的影响
3 结论和展望
在阳光照射下,以海水为原料制取H2是解决能源问题最理想的方法之一。光电催化海水制氢是在光电催化纯水的基础上,结合当前资源状况和能源需求拓展出来的研究项目,旨在利用丰富的海水资源和太阳能转化为清洁的氢能,并缓解淡水资源匮乏的现状。本文综述了至今探索过光电催化分解海水制取H2的研究工作,并对研究内容和催化机理进行了梳理。
催化剂体系以TiO2系为主,大部分作为光阳极使用;研究采用自然光或模拟太阳光源较多,研究体系选取天然海水和浓缩海水为主,对研究NaCl 溶液分解的工作,是以研究反应机理为主要目标;性能数据分布很宽,析氢速率从1.5 μmol·cm-2·h-1到277 μmol·cm-2·h-1不等,这取决于所采用的反应体系与条件,但总体上产氢速率偏低。
目前光电催化海水的研究开展还不够系统深入,大部分工作是科研人员研究光电催化纯水制氢的同时附带的研究内容,并非以海水分解为主要研究方向;在使用的反应环境方面,应尽量选择接近实际的条件,如自然光,天然海水或海盐溶液。多数研究为方便统一,采用了AM 1.5 G为光源,尽量接近自然光。然而,AM 1.5 G的特性与自然光相距较大,催化剂在两种环境下的性能差异很明显。因此建议使用自然光进行材料性能考察;另外,不同文献给出的活性数据不同,建议研究过程中完善数据,增加不同研究的可比性;大部分文章中没有提及稳定性这一催化剂的重要性能,说明光电催化海水材料的稳定性不足,还需进一步增强;最后,由于海水成分复杂繁多,对光电催化海水分解机理的研究明显不够深入全面。开展海水中主要成分对光催化剂及催化过程的影响,这是提升催化效率的基础。与光电催化纯水相比,光电催化海水分解制氢存在两点劣势:(1)由于海水中的成分多且复杂,在反应之前需要过滤海水中的不溶物;(2)海水中不同的离子对不同光电极的作用效果需要具体分析,尚无普适性规律。
在总结的工作中,不乏具有优秀光电催化性能的材料,以及完整且接近实际条件的研究内容,获得了出色的产氢性能。目前,光电催化海水产氢研究已经引起科研人员广泛关注,取得良好进展。利用光伏和海水资源,获取丰富的洁净氢能,是解决能源危机的重要途径之一,我们相信该领域势必取得更大进展。