



1 引言
随着全球经济的不断发展,人类对能源和材料的需求日益增加。在传统化学工业中,化学中间体、聚合物的生产都依赖于化石燃料衍生的石化产品。这些石化产品的生产不但造成了日益严重的环境问题,而且随着化石能源逐渐枯竭,其储量已不能满足工业生产中大量化学品与材料的需求。因此,寻找可以作为化石能源替代品的可再生、可持续的原料迫在眉睫[1]。生物质来源于自然界,储量丰富、分布广泛,可用于生产生物基化学品,因此受到研究人员的广泛关注。以纤维素为例,纤维素是植物细胞的主要结构成分,将结晶纤维素解聚为葡萄糖,可以进一步制得5-羟甲基糠醛(5-hydroxymethylfurfural, HMF)[2],整个转化过程在捕获利用CO2、H2O和阳光的同时释放出HMF、H2O和O2,转化效率高,且全程无有害产物,符合环境友好的生产理念[3]。HMF的生产原料易得,制备路径简单,更重要的是,从HMF出发不仅可以制备各种高价值的化学品,如乙酰丙酸(levulinic acid, LA)[4]、2,5-呋喃二甲醇(2,5-dihydroxymethylfuran, DHMF)、2,5-呋喃二甲醛(2,5-diformylfuran, DFF)[5]和2,5-呋喃二甲酸(2,5-furandicarboxylic acid, FDCA)[6],还可以生成液体燃料替代品,如2,5-二甲基呋喃(2,5-dimethylfuran, DMF)[7]、乙酰丙酸乙酯(ethyl levulinate, EL)[8]、5-乙氧基甲基糠醛(5-ethoxymethylfurfural, EMF)[9]和长链烷烃(long chain alkane, LLA)[10]等。因此,HMF被认为是生物质转化过程中一个非常重要的平台化合物,同时也是联结生物质资源与石油工业的桥梁。
在诸多的糠醛衍生物中,由HMF选择性氧化得到的FDCA尤其引人注目,被认为是化学工业与医药生产中最重要、最有发展前途的分子之一[11]。HMF与FDCA的基本物理性质如表1所示。FDCA的用途广泛,以合成绿色聚合物为例,FDCA具有与石油衍生的聚合物单体对苯二甲酸(terephthalic acid, TPA)相似的分子结构,FDCA可以用于生产聚2,5-呋喃甲酸酯(polyethylene furandicarboxylate, PEF),由FDCA衍生的PEF被认为是石化产品对苯二甲酸衍生物聚对苯二甲酸酯(polyethylene terephthalate, PET)[12]的有效替代物。利用FDCA作为原料,可以有效减少温室气体的排放,并大大降低生产成本[13]。此外,FDCA还可用于生产聚酰胺和增塑剂等多种化学品[14]。
表1 HMF与FDCA的基本物理性质Table 1 Basic physical properties of HMF and FDCA |
| Items | HMF | FDCA |
|---|---|---|
| Molecular weight | 126.11 | 156.09 |
| Boiling point/℃(760 mmHg) | 291.5 | 419.2 |
| Melting point/℃ | 32~35 | 342 |
| Density/g·cm-3(20 ℃) | 1.290 | 1.604 |
| Refractive Index(nD/20 ℃) | 1.562 | 1.461 |
| Flash point/℉ | 175 | 168 |
鉴于FDCA具有优良的理化性质、重要的应用价值和广阔的市场前景,如何通过HMF得到具有更高化学价值的FDCA,是近年来研究人员关注的热点。本文首先归纳各类HMF催化氧化方法中共同存在的反应机理与影响因素,在此基础上,分别对不同催化模式领域中现有研究成果进行总结,阐述了HMF选择性氧化生成FDCA的研究现状及最新进展,并对研究前景进行了展望,以期为寻找一种绿色环保、低能耗高效率的选择性氧化体系提供一定的思路和参考,并为FDCA的规模化生产提供一定的理论依据和技术支撑。
2 HMF选择性氧化制备FDCA的反应机理
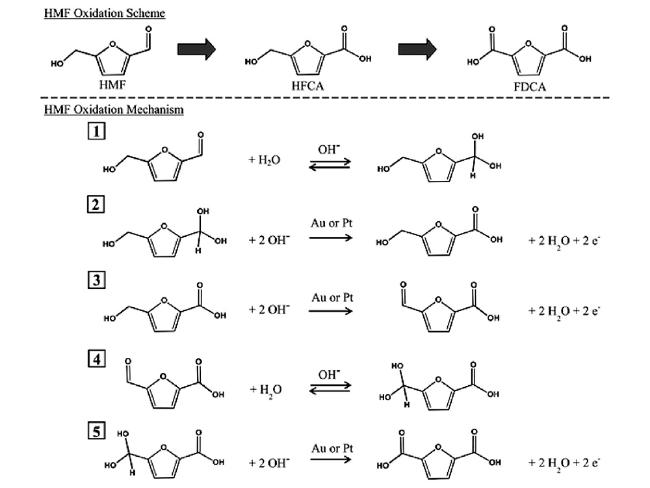
HMF选择性催化氧化生成FDCA是HMF转化利用的一种重要的方式。然而,实现这种转化并不能一蹴而就,如图1所示,根据催化体系的不同,HMF在氧化过程中会经过两种可能的氧化途径,但所有催化体系均需要经过3个步骤才能实现连续氧化HMF分子中的羟甲基及醛基的过程,最终得到含有两个羧基的FDCA[15]。因此,在HMF的连续氧化过程中,除了会生成目标产物FDCA以外,还会产生DFF、5-羟甲基-2-呋喃羧酸(5-hydroxymethyl-2-furancarboxylic acid, HMFCA)、5-甲酰基-2-呋喃羧酸(5-formyl-2-furancaboxylicacid, FFCA)等中间产物。在上述转化过程中,HMF通常能快速转化为DFF或HMFCA,而由FFCA向FDCA的转化则是整个连续反应过程中的决速步骤[16]。研究中发现,FFCA作为中间产物不断积累达到峰值后,将进一步转化为FDCA。
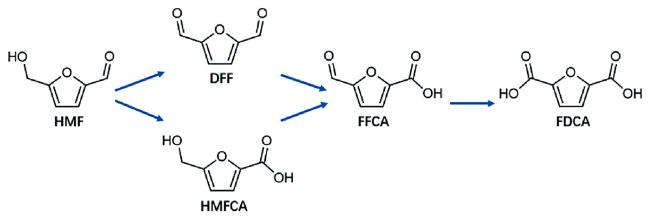
此外,由于HMF中的呋喃环化学性质活泼,在多种活性物质作用下呋喃环自身容易断裂导致开环反应,形成多种副产物,如乙酰丙酸与甲酸(formic acid, FA)等[17]。中间产物与副产物的出现成为FDCA低选择性的主要因素。
3 HMF选择性氧化制备FDCA的影响因素
3.1 氧化剂的选择
在早期的均相HMF(液相-液相)转化体系研究中,研究人员使用KMnO4作为氧化剂[21],得到的FDCA产率较低,且会产生大量重金属废物造成环境污染。在后期的研究中,一些较为绿色环保的液氧源[19],如H2O2,因其具有储存安全、活性氧含量高等优点,开始在工业生产中被广泛应用。近年来,利用H2O2作为氧化剂对生物质进行转化研究屡见不鲜,例如其可将糠醛氧化为马来酸和富马酸[22]。同样作为液氧源,t-BuOOH作为氧化剂可与均相(如CuCl2,FDCA产率45%[20])或异相(如MnFe2O4,FDCA产率85%[23])催化剂组成催化氧化体系,且在两种体系中均展现出较H2O2更高的FDCA产率。但t-BuOOH在水中的溶解度较低,作为氧化剂需应用于极性非质子溶剂(如二甲基亚砜[24]和乙腈[20])体系。然而H2O2与t-BuOOH作为氧化剂存在的共同缺点是反应产物的选择性难以得到控制。例如,当无共催化剂存在时,在反应体系中直接引入H2O2会导致反应底物迅速分解[25]。因此,如果采用液氧源作氧化剂,如何控制氧化剂的浓度或加入量,并选择具有良好循环性的催化剂仍然是设计反应体系时需要考虑的难题。
使用O2作为绿色氧化剂来进行HMF的氧化转化逐渐成为生物质转化应用的主要方式之一。然而,不同于均相反应的是,在涉及液体-气体的两相HMF氧化反应中,O2在反应溶液中的溶解度与催化剂对于O2的活化作用密切相关,因此反应介质对氧气的溶解度将直接影响反应效率。我们将在下一小节对其进行详细阐述。
3.2 反应介质的选择
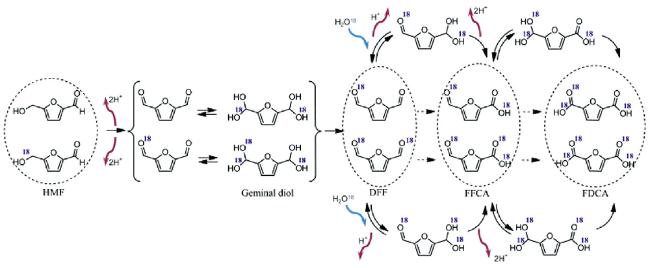
异相催化反应体系中使用的介质通常包括有机溶剂与水溶剂,其中水溶剂应用更为广泛。为理解氧气作为氧化剂与水作为溶剂参与HMF氧化的机制,Davis等[26]用Au/TiO2作催化剂,利用同位素标记技术对HMF氧化产物进行了研究。研究表明,在碱性水溶液中,使用标记后的18O2作为氧化剂时,没有在产物分子中发现18O同位素。而当使用标记水$(H_{2}^{18}O)$作为溶剂时,在相应的产物中检测到18O同位素的存在,结果表明产物中的氧原子来源于溶剂水,而非氧化剂(O2)。氧气在这一转化过程中可能并未直接参与氧化反应,而是起到了转移电子,间接地增强了氧化反应的作用。
尽管有机溶剂能提高氧气的溶解度,但是成本较高且大多具有毒性,可能会对环境造成不可逆的破坏,因此各种催化反应往往更希望能够在水溶剂中进行。不过,溶剂效应对生物质平台分子的催化转化影响显著[28],且已有研究证实将有机溶剂应用在生物质分子的催化转化过程中具有优越的性能[29],是能够提升FDCA产率的重要因素之一。综合以上背景,结合两种溶剂的优点,采用有机溶剂与水溶剂混合的体系来进行HMF转化被认为是行之有效的手段之一。例如,Liu等[30]采用纯水与二甲基亚砜(DMSO)混合作为溶剂,与纯水或纯DMSO相比,DMSO/H2O混合体系能够在碱性环境下稳定HMF,从而实现HMF在高浓度下的高效氧化。Tsilomelekis等[31]通过计算发现DMSO与HMF的结合比水更强,HMF分子中的C  O基团在DMSO/H2O体系中被DMSO优先溶剂化,提高了HMF的LUMO能级,降低了它对亲核攻击的敏感性,从而减少了低价值的副产物生成。这一结果以及DMSO对HMF的优先溶解解释了DMSO/H2O混合体系中HMF稳定性的增强。这些研究结果证明,有机溶剂与水的混合体系避免了大量有机试剂的使用,同时能够实现对反应速率和产物选择性的提高,为研究通过对溶剂的改变提高FDCA产率提供了新思路。
O基团在DMSO/H2O体系中被DMSO优先溶剂化,提高了HMF的LUMO能级,降低了它对亲核攻击的敏感性,从而减少了低价值的副产物生成。这一结果以及DMSO对HMF的优先溶解解释了DMSO/H2O混合体系中HMF稳定性的增强。这些研究结果证明,有机溶剂与水的混合体系避免了大量有机试剂的使用,同时能够实现对反应速率和产物选择性的提高,为研究通过对溶剂的改变提高FDCA产率提供了新思路。
O基团在DMSO/H2O体系中被DMSO优先溶剂化,提高了HMF的LUMO能级,降低了它对亲核攻击的敏感性,从而减少了低价值的副产物生成。这一结果以及DMSO对HMF的优先溶解解释了DMSO/H2O混合体系中HMF稳定性的增强。这些研究结果证明,有机溶剂与水的混合体系避免了大量有机试剂的使用,同时能够实现对反应速率和产物选择性的提高,为研究通过对溶剂的改变提高FDCA产率提供了新思路。3.3 催化剂对反应底物的吸附作用
催化剂对反应底物的吸附模式影响各类异相催化反应,无论是热、电或光催化都需要考虑这一关键因素。以碳基材料负载贵金属为例,Sang等[32]研究发现,采用石墨碳负载Au纳米粒子作为催化剂时,碳基材料表面的羰基官能团促进了HMF在其表面的吸附,同时,Au表面的羟基基团可以减少对FDCA或其他中间产物的吸附,避免活性中心被占据。Wan等[33]将Au-Pd合金负载在经过预处理的碳纳米管(carbon nanotube, CNT)表面,在水中无需额外添加碱(T=373 K, =0.5 MPa)即可得到94%的FDCA产率。他们认为,经H2O2预处理后碳纳米管的表面功能化是提高FDCA产率的关键:当碳纳米管表面含有较多羰基时可以促进其对反应物和反应中间体的吸附,而表面有较多羧基时则会起到相反的作用。
调控表面基团是改变催化剂对反应底物的吸附行为的关键,对于不同材料调控方法不尽相同。以碳材料与金属氧化物为例,调控碳材料表面基团已有大量研究工作,例如通过化学氧化法(KOH[36]、HN 、H2O2等)或电化学氧化法[38]能够在材料表面引入大量含氧基团。表面含氧基团种类丰富,通常包括羟基、羰基、羧基或与两个碳原子形成醚键等[39],通过热处理[40]或将化学与电化学氧化相结合[41]等方式能够有效调控碳材料表面的含氧基团数量并选择性转化部分基团,是调控基团以获得最佳吸附状态的有效手段。对于金属氧化物,掺杂是改变材料表面基团数量与亲疏水性的重要方法,以CeO2为例,Megías-Sayago 等[42]在CeO2中引入Zr元素,制备不同Ce/Zr摩尔比的CexZr1- x O2载体,并负载Au催化剂,FDCA的产率与实验观察到的羟基数量线性相关。Zr的参与,为催化剂表面带来更多的羟基基团,有利于呋喃环上羟甲基的吸附并加速其脱质子过程,有效提高了HMFCA向FFCA的转化速率。
综上,催化剂与反应底物之间的吸附作用对催化反应最终所获得的产率影响显著,但在当前的相关研究中研究人员对催化剂表面与底物的相互作用的讨论仍然不足,在未来的研究中通过调控催化剂表面基团进一步提高催化剂性能是不可忽视的重要一环。
4 热催化氧化反应
4.1 贵金属催化剂
利用贵金属材料进行热催化氧化HMF来获得高选择性的FDCA产物是目前研究最为广泛的材料体系之一,其中钌(Ru)、铂(Pt)、钯(Pd)和金(Au)是最常用的催化材料。在实际应用中,通常需要将贵金属负载在碳化物或氧化物载体上进行催化氧化反应。载体的表面性能以及金属和载体之间的相互作用在很大程度上影响催化材料的最终性能,因此载体的选择至关重要。
目前的HMF氧化反应研究中,最为常见的是以碳材料作为载体,负载贵金属颗粒作为催化剂。碳材料具有较大的比表面积、高导电性以及良好的化学稳定性,作为载体具有原料易得、价格低廉的优点,更重要的是碳有多种多样的结构形式,可以人为地调控其形貌来满足不同的负载需求。Yi等[43]研究了在无碱水溶液中,利用Ru/C催化剂将HMF催化转化为FDCA,产率为88%。类似地,Chen等[19]采用活性炭(activated carbon, AC)负载钌(Ru/AC)为催化剂,H2O2为液氧源,合成FDCA的产率可达91%。在该研究中,他们利用各中间体的反应速率不同,通过调整反应底物HMF与所用催化剂的比例,可以选择性地控制最终反应产物为FFCA或FDCA。
除了负载单一贵金属,贵金属合金因其显著的协同效应,在提高HMF转化率和FDCA选择性方面体现出独特的优势。Villa等[44]报道,负载在活性炭上的Au-Pd合金纳米颗粒可以催化氧化HMF为FDCA。催化剂Au8Pd2/AC(其中Au和Pd的比例为8∶2)具有较高的催化活性,在相对温和的条件下(T=333 K),HMF可获得大于59%的转化率。Wan等[33]通过如图3a所示的对比实验,发现CNT负载的Au单金属催化剂将优先催化HMF中醛基的氧化,在反应的前期得到HMFCA,而随反应时间的延长,在无碱条件下则发生开环和副产物降解反应。在图3b中CNT负载Au-Pd合金纳米粒子通过加速HMF中羟基的氧化,能够改变反应的主要路径,得到目标产物FDCA。由此可见,负载的Au-Pd合金也是实现选择性生成FDCA的关键因素,同样使用CNT负载的单金属Au或Pd作为催化剂得到的转化率和选择性远低于负载合金的性能。
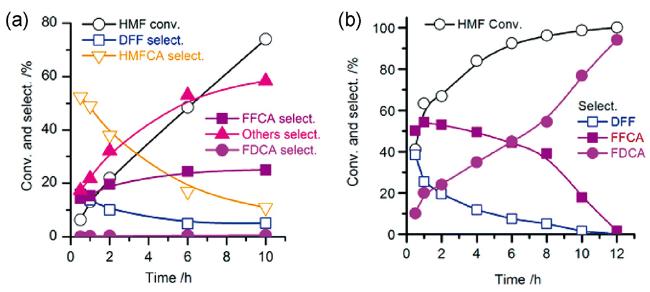
图3 (a) HMF在Au/CNT催化剂上转化的过程;(b) HMF在Au-Pd/CNT催化剂上转化的过程;(反应条件:HMF为0.50 mmol;HMF/Au(摩尔比)200/1;H2O 20 mL; O2 0.5 MPa; 温度373 K)[33]Fig.3 (a) Time course for the conversion of HMF over the Au/CNT catalyst;(b) Time course for the conversion of HMF over the Au-Pd/CNT(Au/Pd=1/1) catalyst(Reaction conditions: HMF, 0.50 mmol; HMF/Au(molar ratio), 200/1; H2O, 20 mL; O2, 0.5 MPa; temperature, 373 K)[33] |
除碳材料外,贵金属催化剂还使用其他材料作为载体,常见的有树脂、PVP、CeO2与TiO2等。Antonyraj等[45]利用碱性阴离子交换树脂(anion-exchange resin, AER)负载Au-Pd合金纳米颗粒对HMF进行氧化,5~20 nm的Au-Pd合金纳米粒子能够在树脂表面和树脂球内均匀分散。与前文类似地,他们对在AER载体上以合金形式存在的Au、Pd,与AER负载的单金属纳米粒子进行了比较研究(T=373 K, =1 MPa)。负载合金时,HMF被氧化成FDCA的产率为93.2%;而负载单金属并混合作为催化剂时,在相同条件下仅能获得52%的产率,实验结果进一步验证了贵金属Au-Pd合金对HMF存在的协同催化效应。
4.2 过渡金属催化剂
以贵金属作为催化剂的研究已经非常全面,但贵金属高昂的价格、地球资源的稀缺等限制了其进一步发展与应用。在这种前提下,过渡金属元素在催化领域开始得到关注,与贵金属相比,过渡金属具有储量丰富,价格低廉等多种优点。Hayashi等[47]利用MnO2和NaHCO3构建催化剂反应体系(T=373 K, =1 MPa),在这个反应体系中决速步骤的反应速率根据MnO2结构的不同而存在差异,因此HMF氧化生成FDCA的转化效率在很大程度上取决于MnO2的晶体结构。Han等[48]采用共沉淀法制备MnO x -CeO2混合氧化物催化剂,当使用MC-6(Mn/Ce=6)催化剂时(T=348~383 K, =2 MPa),催化转化FDCA的产率可达91%。这一结果表明过渡金属氧化物在水溶剂中催化HMF氧化为FDCA也具有优异的性能。通过对催化剂结构的进一步分析表明,在不含贵金属的情况下,表面Mn4+可以作为HMF氧化的活性中心。此外,非贵金属元素间同样存在着协同效应,即Mn与Ce氧化物的协同作用,有助于提升催化剂的催化活性。利用非贵金属氧化物协同作用的还有Ventura等[49],他们报道了一种CuO·MnO2·CeO2的混合氧化物,在无碱条件下能将HMF转化为FDCA,产率为71%。反应过程中,Cu(Ⅱ)被还原会导致催化剂的暂时失活,但将催化剂在823 K的温度下重新焙烧处理后,可以实现FFCA氧化为FDCA的催化过程,提高FDCA产率至99%。
4.3 非金属催化剂
除了基于金属催化剂的均相与非均相催化体系外,在近年的研究中,非金属催化剂的出现扩大了催化剂的选择范围,受到越来越多研究人员的关注。与过渡金属催化剂相比,非金属催化剂成本更加低廉,处理与回收过程环保。此外,金属离子从金属催化剂中浸出到液相反应体系中会不可避免的造成反应产物提纯受阻。因此,非金属催化剂被看作为未来具有广阔研究前景的催化剂体系。
在针对HMF的催化氧化反应中,以碳与氮为基本组成元素的非金属催化剂展现出卓越的性能。例如,氮掺杂碳材料(NCS),具有制备工艺简单、成本低、催化性能好等优点,在电化学、催化等领域引起了广泛的兴趣。Nguyen等[16]通过合成含氮的沸石咪唑骨架(ZIFs),经过热处理后,去除金属离子并使骨架碳化,同时在骨架中保留氮,制备出含氮纳米孔碳(NNC)材料,这种非金属催化剂可以达到80%的FDCA产率(T=373 K),但完全转化需要48 h。Verma等[50]在此基础上进一步降低了反应所需温度并缩短了反应时间,通过在氮气气氛下煅烧壳聚糖得到的多孔氮化碳催化剂(PCN x ),可以在36 h内得到83%的FDCA产率(T=373 K)。
与金属催化剂不同的是,非金属催化剂NNC与PCN x 上的石墨化氮被认为是催化活性位点,可以将氧气活化为具有高氧化性的氧自由基[16]。他们通过添加活性氧猝灭剂并对比不同石墨氮含量的催化剂验证了石墨氮位点对氧气的活化作用。需要注意的是,不含石墨氮活性位点的非金属催化剂,如石墨、石墨烯、碳纳米管等碳材料,在相同的实验条件下,不具备催化氧化HMF生成FDCA的能力。这些研究说明石墨化氮活性位点的建立与调控是建立非金属催化体系的研究重点。
4.4 添加碱的作用机理
加入弱碱具有同强碱性物质类似的作用机理,能够促进HMF向FDCA的转化[48],并且可以提供适中的pH环境,减少反应底物的降解[51]。除此之外,弱碱的添加能够促进中间产物的溶解,避免生成的FFCA或其他中间产物吸附在催化剂活性位点上,从而保持催化剂的活性。在没有额外添加碱的情况下,利用材料自身的固体碱催化位点也可以催化HMF的氧化,但是所得到的中间产物如DFF与FFCA可能会占据催化剂表面,阻碍中间产物进一步的氧化。例如,Ventura等[52]报道了直接采用一种铜与铈氧化物(CuO·CeO2)固体碱催化剂,在没有额外添加碱性物质辅助的情况下进行HMF选择性氧化反应(T=383 K, =0.9 MPa)时,获得的主要产物为FFCA,即使增加反应时间也不能进一步氧化得到FDCA。
如前文所述机理只在碱性物质存在的情况下可以实现,而无碱添加的HMF氧化则可能是另一种反应机制。Siankevich等[27]通过同位素标记证实了在不添加碱性物质的条件下,氧化反应所经历的路径是由DFF至FFCA再最终氧化为FDCA,而不同于碱性条件下经由中间体HMFCA的氧化过程。反应过程类似于碱促进氧化第1步,水与醛基发生亲核加成反应,迅速产生偕二醇,之后通过分子氧与催化剂材料表面作用,实现质子的迁移得到羧基,从而使HMF实现无碱氧化过程。
综上,在贵金属或过渡金属催化剂上进行HMF选择性热催化氧化(T=333~423 K)已经有了广泛的研究和认识,产率高与产物纯是热催化氧化的突出优势,但仍存在包括贵金属催化剂成本高、能耗高(反应温度高)、氧压高等缺点,在一定程度上阻碍了其在工业中的发展。此外,碱的额外添加也不利于产物的分离,同时对环境造成了负担。在热催化的未来研究中,构建无需添加碱仍能够高效、高选择性氧化的反应体系,无疑会成为研究的热点与挑战之一。
5 电催化氧化反应
电化学HMF氧化是通常在常温下进行,其氧化过程受阳极电位的驱动,能够避免额外添加化学氧化剂,环境友好。电催化氧化反应阴极与阳极同时参与反应,即OER过程与HER过程同时进行,当阳极OER过程被HMF的氧化过程代替时,由于HMF氧化在热力学上比水氧化更有利,同时使H2的产生效率也能够得到提高。随着可再生能源发电(如太阳能和风能)的成本持续下降,电催化的成本竞争力也将日益增强[53] 。而目前电化学HMF氧化研究的主要问题在于,大部分电极还是由成本较高的贵金属(Pt,Au,Ru,Pd)组成,并且对期望产物FDCA的选择性较低。例如Chadderdon等[54]研究了碳负载Au和Pd纳米粒子在碱性介质中对HMF的电催化氧化。结果表明,HMF(20 mM)可在0.9 V(vs RHE)下在1 h内完全氧化,但FDCA的最高选择性仅为83%。
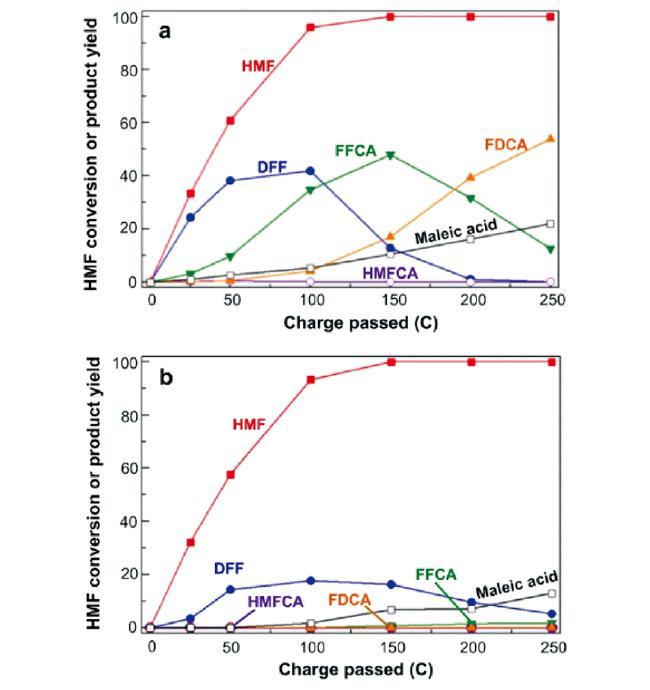
图5 在1.6 V(vs RHE)下使用MnO x 阳极(a)与在2.0 V(vs RHE)使用Pt阳极(b)的HMF随时间变化的转化率与各产物选择性(反应在含20 mM HMF的pH=1的H2SO4溶液进行)[56]Fig.5 Conversion of HMF(%) and yield(%) of oxidation products obtained by using MnO x anodes at 1.6 V(vs RHE)(a) and Pt anodes at 2.0 V(vs RHE)(b) during the course of electrochemical oxidation of HMF in a pH=1 H2SO4 solution containing 20 mM HMF at various amounts of charge passed[56] |
同时,HMF氧化反应的路径受到电极种类与外加电压的影响。Vuyyuru等[55]以Pt为工作电极,研究了电解液pH值对HMF电催化氧化的影响,在强碱性条件下(pH>13)HMF稳定,当电流密度为0.44 mA·cm-2、pH=10时,HMF的转化率约为70%。然而,产物中仅获得了18%的DFF,FDCA的数量可以忽略不计(<1%),大多数的氧化产物未能确定。同样,Choi等[56]发现MnO x 阳极可将HMF的羟基和醛基氧化成羧基,形成FDCA,如图5所示。而在相同条件下,使用Pt作为阳极,可选择性氧化羟甲基形成DFF,但不能进一步氧化DFF为FDCA。此外,使用Pt作为电极还会促进HMF生成胡敏素,阻碍了反应底物向更有价值的产品的转化。Cao等[57]在Pt-Ru电极上,在323 K温度下通电氧化17 h,合成了DFF(产率为89%),而FDCA的产率极低。综合贵金属的价格与其性能,Pt并不是HMF选择性氧化为FDCA的有效电催化剂[58, 59]。
如前文所述,贵金属作为电催化剂的性能并不突出,且高昂的成本也制约了它的发展,与热催化相似,人们将过渡金属催化剂视为催化HMF氧化的良好替代材料。镍基催化剂首先得到研究人员的注意,早在1991年Grabowski等[60]首次报道了HMF的电化学氧化方法制FDCA,在NaOH(1.0 M)溶液中,使用NiO/Ni(OH)2阳极实现选择性转化,FDCA产率为71%。双金属催化剂可以利用协同效应进一步提升催化性能,Kang等[61]研究了尖晶石结构Co基金属氧化物电极对HMF氧化的活性,采用简单的水热法在泡沫镍上生长了丝状纳米结构的Co3O4和NiCo2O4电极,其中NiCo2O4对HMF转化率可达99.6%,对FDCA的选择性为90.8%。如图6所示,同样使用Ni-Co过渡金属氧化物的还有Gao等[62],他们在报道中比较了不同Ni与Co比例的NixCo3-xO4的形貌结构与性能,其中以NiCo2O4(Co/Ni=2∶1)作为催化剂用于HMF的电催化氧化得到的性能最佳,FDCA产率为90%,法拉第效率接近100%。Ni-Co基过渡金属氧化物作为一种非贵金属催化剂,被看作高效的电化学催化氧化HMF的电极材料。此外,Weidner等[63]研究了各种钴-金属合金(CoX;X=B,Si,P,Te,As)作为HMF氧化催化剂的性能,在所测试的一系列材料中,CoB是对催化HMF氧化反应最活跃的电催化剂,在1.45 V(vs RHE)的电压下,HMF的转化率与FDCA的选择性均接近100%。

值得注意的是,Choi等在使用过渡金属作为催化剂电催化氧化HMF到FDCA的方面做了一系列的研究工作。除了如前文所述对MnO x 性能进行了探讨,还对Cu[64],NiOOH与CoOOH[65]进行研究。他们的研究表明,以不同形态的Cu为催化阳极,例如纳米晶铜电极和块体铜电极,对HMF电化学氧化制FDCA有较大的影响。虽然两种电极都能够实现HMF向FDCA的转化,但纳米晶铜电极在氧化过程中,电极表面形成了非晶态氢氧化物和氧化物,其中含有特殊配位环境的Cu离子,与HMF有很强的相互作用,有利于HMF的氧化过程。在此之后,Choi等又对NiOOH和CoOOH对HMF催化氧化性能进行系统的研究。制备了不同厚度的MOOH(M=Ni,Co)薄膜,比较这些材料在KOH(0.1 M,pH=13)溶液中的催化活性。实验证明,NiOOH是最有效的电化学氧化催化剂,在1.47 V(vs RHE)下,其FDCA产率可达96.0%,而使用Co(OH)2/CoOOH作为催化剂,不能产生足够的电流密度使HMF在恒电位下氧化,导致氧化速率较慢。
对电催化剂的改进无疑是突破电催化氧化瓶颈重要的一环。在使用相同的基底时,催化剂的结构设计对于性能提升尤为关键。近期Gao等[66]报道了一种NiSe@NiO x 核壳纳米线结构的电催化剂:以NiSe作为高导电性框架,表面NiO x 层提供反应活性位。这种独特的结构暴露出更多的活性位点,能够有效提高FDCA的产率。
除了对于催化剂本身进行改性优化之外,在最近的电催化氧化研究中,有研究人员将太阳能与电催化相结合,以期达到自然能源的最大利用。Li等[67]利用太阳能电池(<1.7 V)进行电催化水分解与HMF氧化相耦合的研究。通过太阳能电池直接提供阴阳两极的反应过程所需的外加电压,在接近中性的溶液中,阴极反应过程可以产生氢气,而阳极反应过程则实现HMF向FDCA的氧化转化过程。综上,将HMF的电化学氧化生成FDCA与阴极还原产氢反应相耦合,是提高电催化反应效率的一种新途径。
6 光催化氧化反应
从前期的研究中可以看出,反应中产物选择性低的原因是由于在反应中生成了氧化能力过强的活性氧物种如羟基自由基(·OH),其在氧化过程中会破坏呋喃环自身结构,造成大量开环产物降低了反应的选择性[74]。因此在针对光驱动的HMF催化氧化过程中,对于活性氧物种的选择与调控至关重要。目前,单线态氧(1O2)是一个备受关注的重要的活性氧物种,单线态氧是氧分子在活化的过程中,由催化剂中三线态激子与基态氧分子之间的发生共振能量转移而产生的[75]。单线态氧具有独特的电子结构和较强的氧化能力,常常应用于处理污染物,同时在生物与医学等领域也有着重要应用[76, 77]。光催化剂活化分子氧所产生的1O2,可将醇氧化成羰基化合物,或将醛氧化成羧酸。例如,Iqbal等[78]报告了在Ru或Ir光催化剂存在下,由可见光产生的1O2可以将醛定量氧化为羧酸,并且对于多种含醛有机物,生成相应的羧酸选择性都可以达到90%以上。
近期的研究表明,通过Ru这样的贵金属作为催化剂可以产生1O2。虽然这种催化方式具有优异的选择性,却大大增加了催化反应的成本。在目前的研究中,人们期望寻找能够兼具选择性产生1O2,且价格低廉、环境友好的催化剂[79, 80]。其中,石墨相氮化碳(g-C3N4)因为具有表面性质丰富、热稳定性和化学稳定性好、电子结构可调等优点,在催化领域广受关注,已有多种方面的应用研究[81]。金属硫代卟啉具有一些独特的能级结构和氧化还原特性,可以被用于光催化降解有机污染物。如图7所示,Xu等[82]制备了CoPz/g-C3N4,CoPz(钴配位硫代卟啉)在被光激发后通过能量传递将三线态氧3O2转化为单线态氧1O2,进而氧化HMF到FDCA,HMF转化率可以达到99.1%,FDCA选择性为97%。他们在研究中证明1O2可选择性地将HMF氧化成理想的FDCA。并且,他们提出反应溶液的pH值是影响HMF氧化的一个重要因素,会影响到反应终产物的种类。当利用Na2B4O7调节反应溶液的pH=9.18时,产物主要为FDCA,而在低pH(pH=4.01)条件下,主要产物为氧化中间体DFF。
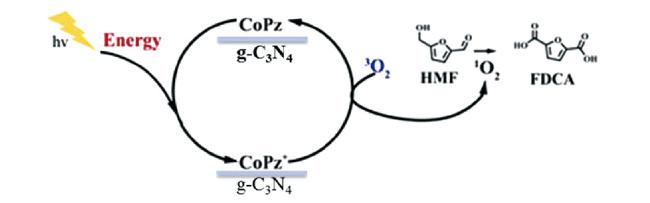
对半导体光催化剂表面进行改性是调控氧自由基种类与数量的有效手段之一,例如,Ilkaeva等[83]通过H2O2修饰g-C3N4催化剂表面,能够减少氧化能力过强的羟基自由基生成。g-C3N4边缘因为具有端氨基等基团,成为布朗斯特碱性位点,与水分子之间形成氢键,在催化剂表面产生-N 基团。在这种相互作用的状态下,光生空穴攻击水分子将产生大量羟基自由基。然而,当H2O2与表面端氨基之间相互作用后,H2O2占据端氨基位点,水分子难以解离为羟基再参与反应过程。因此,在表面修饰H2O2后,光照激发下g-C3N4能够选择性的产生氧化能力适中的超氧自由基,应用于高选择性的光催化过程中。
光能是研究生物质平台分子转化过程中不可忽视的能量来源,光催化能够直接利用太阳光,具有可持续发展的特点。与热催化和电催化相比,利用光催化技术来进行HMF转化的相关研究仍然较为缺乏,活性氧物种的调控是决定产物选择性的重要因素。尤其是在低毒性低污染性的溶剂体系中实现活性氧物种选择性生成的详细机理,仍然需要深入研究。
7 生物催化氧化反应
近年来,使用生物方法催化氧化HMF逐渐受到关注,与化学催化氧化反应相比,生物催化转化法的反应条件较为温和,且不需要采用有毒的化学品,提供了一个环境友好的反应体系。生物催化氧化HMF通常使用酶作为活性物质,在较低的环境温度/压力与生理pH等温和的反应条件下,不同的酶会产生不同的催化效果,选择性地生成相应的HMF衍生物,减少了副产物的形成。
早在1997年,Marion等[84]提出了氯过氧化物酶能够氧化HMF,但在研究中HMF未能被氯过氧化物酶完全氧化,产生的是DFF(65%)与HMFCA(35%)的混合物。类似的,Zhang等[85]从枯草芽孢杆菌(Bacillussubtilis TJ-102)中筛选出一种漆酶(Cota-TJ 102),漆酶具有较为温和的特性,能够选择性地将HMF转化为FFCA,但不能进一步氧化为FDCA。一般来说,由于酶具有高底物特异性,通过单一酶直接氧化HMF生产FDCA是较为罕见的。因此通常涉及几步氧化反应过程,其中包括分步进行的串联氧化反应与同时使用多种酶进行的一锅催化,均可以实现终产物为FDCA的反应过程。
酶的串联氧化反应针对氧化路径中的中间产物,分别选择合适的酶并进行组合,具有高选择性的优势。Qin等[86]设计了半乳糖氧化酶(GO)和脂肪酶的串联氧化反应。如图8所示,首先,通过GO将HMF转化为DFF,经过48 h后,HMF的转化率达到75%左右。之后采用固定化南极假丝酵母脂肪酶(CAL-B,Novozym 435)和H2O2对DFF进行氧化。第二步反应进行迅速,DFF在7 h内完全转化,开始时以FFCA为主要产物,随着反应时间的延长,中间体完全转化为FDCA,产率为88%。 然而,多种酶之间存在着最适反应条件不同的问题,这为串联反应的设计,反应中间产物的提取都带来了额外的阻碍,不利于产业化生产。Dijkman等[87]发现一种HMF氧化酶(HMFO),属于葡萄糖-甲醇-胆碱氧化还原酶(GMC)族,在较宽的pH范围内具有较高的热稳定性和活性,能够在常温常压下单独进行HMF的三步串联氧化。单一氧化酶HMFO本身为醇氧化酶,对醛的氧化是以氧化偕二醇的形式进行,这降低了FDCA的生产效率。在已有的研究工作中针对HMFO的突变改造,提高了HMFO的热稳定性和有机溶剂耐受性,并部分提高了FDCA的产率[88, 89]。单一酶串联氧化大大降低了催化反应的成本,同时简化反应流程,目前在通过对单一氧化酶的改进提升FDCA生产速率方面仍具有广阔的研究空间。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
利用酶与化学结合的方法在近年逐渐得到研究人员的关注,将酶-化学的组合与串联氧化反应相结合,能够有效利用酶催化的选择性,化学催化的高效性。例如,Krystof等[92]利用化学催化剂和酶串联两步法,首先利用2,2,6,6,-四甲基哌啶氮氧化物(TEMPO)氧化HMF为DFF,再利用脂肪酶氧化乙酸乙酯原位产生的过氧酸将DFF氧化为FDCA,FDCA产率可以达到93%。此外,将多酶一锅氧化的设计思路与酶-化学的催化组合相结合能够补充酶高底物特异性的短板,扩展底物范围。例如漆酶-四甲基哌啶氮氧化物(TEMPO),是经典的酶-化学催化体系,能够选择性的氧化醇,但对于醛基的氧化能力很低。Yang等[93]在使用漆酶-TEMPO的同时一锅加入能够选择性氧化醛的睾丸酮丛毛单胞菌(Comamonas testosteroni SC1588),FDCA产率可达87%。
除了分离酶胞外催化外,全细胞生物催化方法也被用于生产HMF的各种衍生物。全细胞生物催化方法利用完整的生物体作为催化剂,细胞膜的存在为酶创造了一个较为稳定的环境,不易失活,催化稳定性高。同时,全细胞催化无需对酶进行分离纯化,大大降低了催化的时间与经济成本。Koopman等[94]从碱蓬中分离出一种新的HMF氧化还原酶,可以将HMF转化为FDCA。将编码该氧化还原酶的hmfH基因导入恶臭假单胞菌(Pseudomonas putida S12),利用全细胞生物催化剂将HMF转化为FDCA,产率为97%。在Hossain等[95]的研究中,基于细菌可以从HMF中产生FDCA的能力,从土壤中分离出解鸟氨酸拉乌尔菌(Raoultellaornithinolytica BF60)。Yuan等[96]在此基础上通过筛选获得一株可产FDCA的菌株,并且优化hmfH和HMFO在解鸟氨酸拉乌尔菌中的共同表达,以进一步提高全细胞生物催化HMF产生FDCA的能力。
全细胞催化氧化中目前仍存在一定的瓶颈,在之前所报道的多种氧化酶氧化过程中,将1 mol HMF氧化为FDCA需要2~3 mol ,但如前文关于机理章节所述,O2在水中的溶解度有限,全细胞催化中,氧化反应与全细胞自身之间存在O2的竞争,限制了反应的速率。针对O2在水中的低溶解度对生物催化方法所存在的限制,目前氧化酶与脱氢酶偶联可能是能够突破这一瓶颈的发展方向之一,氧化酶氧化形成的副产物H2O2可以实现内循环,从而降低此类反应对O2的需求[97]。
8 结论与展望
基于廉价的5-羟甲基糠醛(HMF)平台化合物,通过选择性氧化反应生成重要的化工与医药生产中间体呋喃二甲酸(FDCA),是今后生物质能源领域最具前景的研究方向之一。目前对于HMF选择性氧化制备FDCA的研究已取得了突出的进展,但期望实现大规模工业制备FDCA来替代传统石化原料,仍存在着较大困难,所采用的催化体系仍具有一些明显的缺陷,亟待在后续研究中解决。在今后的研究过程中,需要特别重视以下几方面的问题。
(1)建立无碱或弱碱反应体系。在HMF选择性氧化反应研究中,一般会向反应体系中引入过量的强碱NaOH或KOH来促进该反应的进行,造成了反应结束后产生大量碱性废液,不能直接进行排放。此外,强碱的引入也对工业生产中的设备提出了更高的要求,增加了设备成本。因此,用高活性的固体碱催化剂来替代强碱,将固体碱催化的优势与其他催化反应模式相结合,使其在中性反应溶液中能获得高选择性的目标产物FDCA。。
(2)氧化反应与析氢反应的耦合。在HMF转化为FDCA的过程中可能伴随着加氧脱氢的过程,但是这个脱氢反应过程中产生的氢气却没有得到很好的利用。利用电催化和光催化反应体系中氧化反应和还原反应耦合的特点,设计合理的反应体系,使脱氢反应与HMF的选择性氧化反应耦合起来,则有可能同时实现氢气利用和FDCA的高效制备。
(3)开发低成本、高活性的催化材料体系。HMF选择性氧化制备FDCA反应的研究中,多采用Au、Pt、Pd和Ru等贵金属作为负载型金属催化剂。虽然这些催化剂可在反应过程中表现出突出的催化活性,但所采用的贵金属催化剂价格较高、分布少,而且贵金属催化剂在热催化过程由于团聚等原因会发生失活,使得催化剂成本极大地提高。因此,开发基于非贵金属的催化体系,通过表面修饰改性或微结构的调整,提高催化剂效率和稳定性对推动其大规模生产应用尤为重要。