




1 引言
随着世界能源的日益紧张和环境的持续恶化,近年来新型清洁能源,如太阳能、风能、潮汐能等开始走向大规模应用。随之而来的能量分布式存储(电动汽车、移动电源等)和调峰蓄谷(大型储能电站)等大规模应用对二次电池的需求量越来越大[1]。虽然锂离子电池由于诸多优点是目前阶段的首选体系,但随着未来需求量的持续增加,锂资源的不足将可能困扰世界。地壳中锂的丰度仅为20 ppm,而同族相邻周期的钠,其丰度高达23 600 ppm,是锂的1180倍。如果用钠代替锂,将完全不需要担心钠的资源问题。因此,自2010年以来,钠离子电池的研究呈现爆发式增长,成为“后锂离子电池”(Beyond lithium-ion batteries)领域的重要分支[2,3,4,5,6]。
虽然锂和钠同属于碱金属元素,但相对于Li+,Na+的离子半径较大(1.02 Å vs 0.76 Å)。这种离子半径的差异,使得钠离子电池和锂离子电池相比,除了资源和价格因素外还有如下优势:首先从结构上,由于Na+与过渡金属离子半径差异远大于Li+与其的差异,这使得困扰锂-过渡金属复合氧化物的离子混排问题难以出现,因此材料更容易合成。其次更大的离子半径意味着在极性溶剂中拥有更低的溶剂化能,这将有利于离子在固液界面的嵌入/脱出,从而有利于获得倍率性能更好的电池。最后,含有Na+的电解液有着更高的离子电导率[3]。综上所述,钠离子电池不仅拥有资源丰富和原料价格低的优点,在材料合成和电化学性能上也都具有相当好的发展潜力,使得钠离子电池在大型应用领域具有很强的竞争力。但是,也由于Na+和Li+离子半径的差异,使得Na+的固相扩散阻碍更大,材料结构和充放电特性也和锂离子电池材料存在一定差异,因此引起了人们极大的研究兴趣。
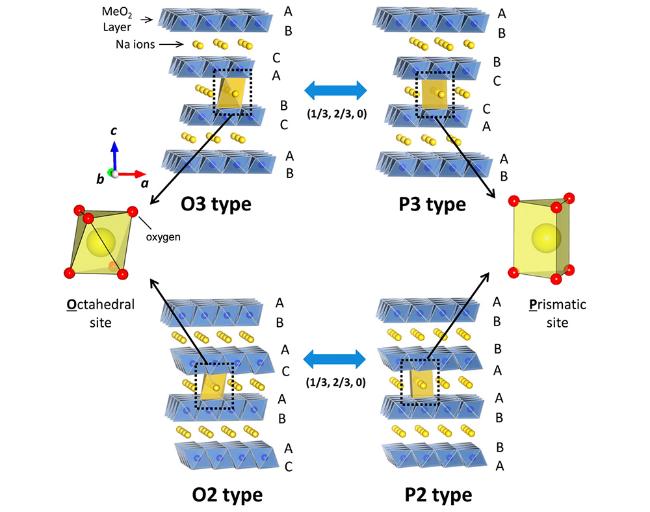
正极材料的容量、电压以及其他电化学性能,在整个电池体系中起着主导地位。研究正极材料的合成、优化和电化学性能,对于钠离子电池的发展具有重要意义。已经发现的钠离子电池正极材料体系有橄榄石结构磷酸盐[7,8,9,10]、氟磷酸盐[11,12,13,14]、NASCICON(Na+快离子导体)化合物[15]、层状过渡金属复合氧化物[16,17,18,19,20,21,22,23,24,25,26]以及普鲁士蓝[27,28,29,30,31,32]等。在这些体系中,层状过渡金属复合氧化物由于合成方便、结构简单和原料来源广等优点,是实用化钠离子电池正极材料的有力竞争者[33]。它们能够提供高于100 mAh/g比容量和3.5 V的电压[34,35]。通常用NaxTMO2表示层状过渡金属氧化物,其中TM代表过渡金属元素,如Co、Fe、Mn、Ni、V、Cr、Ti、Mg等,可以有一种或多种;x则代表Na的化学计量数,范围为0<x≤1[36]。早在1980年,Delmas等[37]就对钠离子电池正极层状材料的储钠机理进行了深入的研究,并将其分为P型结构和O型结构。如图1所示为P型结构和O型结构的结构示意图,其中P和O代表的是不同密堆积层中钠离子处于不同的氧配位环境,P代表三棱柱配位、O代表八面体配位。又可以根据晶胞内氧的最小重复单元堆垛层数将其细分为P2、P3、O2、O3四种结构。以P2结构为例,Na+占据阳离子形成的三棱柱空隙位,且过渡金属层中氧原子的堆剁方式在一个周期中出现两次,呈ABBAABBA…排列。
相对于其他结构的层状材料,P2结构材料具有更高的放电容量[38,39]、优异的倍率性能以及较好的循环稳定性,且在低Na浓度情况下,P2结构材料的离子电导率高于其他几种材料[40,41]。一般情况下,O3结构材料的初始钠含量高于P2结构材料,理论上具有较髙的容量,但是当Na+迁移时需要经过四面体中心位置,扩散势垒较大,因此实际情况下P2结构材料比容量较高,最高可达200 mAh/g,而O3结构材料一般在150 mAh/g以下[42]。同时,P2结构中钠离子是经过一个相对宽阔的平面四边形中心的位置,具有开放的棱柱路径,即有着更大的层间间距,扩散势垒相对更低,具有更快的电子和离子转移速率,因此表现出更突出的倍率性能[43,44,45]。此外,P2结构材料通常是在较高温度下(800 ℃)煅烧而合成的,在发生转变时,需要断开TM—O键,使相变发生较为困难,相较于O3结构材料要更为稳定[33]。所以,综合以上原因,P2结构材料相对于其他材料具有更为明朗的实际应用前景。
2 含单一过渡金属的P2结构材料
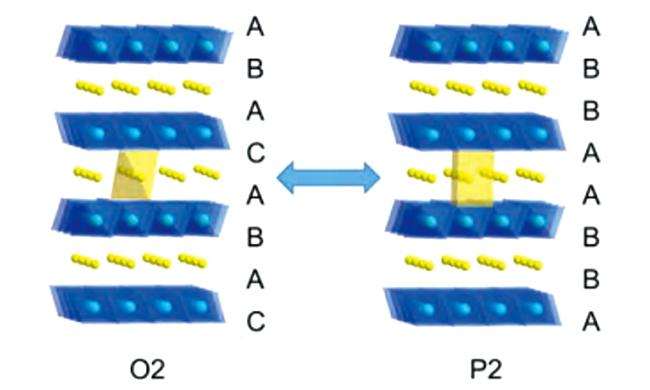
锰系钠氧化物材料(NaxMnO2)材料因其环境友好和丰富的锰矿资源引起广泛关注,当x小于0.45时,可以有规律地形成隧道结构,例如Na0.44MnO2[60]。相比之下,当x大于或等于0.45时,通常会得到层状结构,如层状Na0.5MnO2[61]。Li等[62]通过配合络合沉淀法和后续高温煅烧法制备了层状P2结构Na0.53MnO2纳米棒,该材料在电压范围为2~4.3 V(vs Na+/Na)0.1 C倍率下,可提供高达135.7 mAh/g的高可逆容量(理论容量为140 mAh/g),对应于0.51 mol Na+的嵌入/脱出。在2 C的倍率下,经过50圈循环后,容量仅减少了6.8%,其容量的衰减部分归因于钠离子在反复脱嵌过程中导致的结构不稳定性,但主要原因应是Mn3+的Jahn-Teller效应的影响[63]。当高自旋态的Mn3+发生Jahn-Teller效应时会被歧化成二价和四价锰离子,从而造成不可逆容量损失,这也正是锰系材料存在的固有缺陷[64,65]。P2结构的Na0.7MnO2作为一种高容量、高导电性的钠离子电池正极材料,具有200 mAh/g的比容量和高的体积能量密度。然而,该材料容量在循环过程中衰减非常严重,循环100圈后容量会急剧下降至41.3 mAh/g,对应的容量保持率仅为20.7%,从而限制了其实际应用[66]。其容量衰减与上述Na0.53MnO2存在同样的原因,即在高电压(低Na+含量)下发生的不可逆相变(图2,P2到O2或OP4)以及在特定Na+含量下形成的不同Na+/空位有序模式之间的转变是造成容量损失的关键因素[67]。鉴于此,杨学林等[68]通过碱化-煅烧法将NaxMnO2纳米片阵列沉积于泡沫镍衬底上,得到的NaxMnO2/Ni纳米结构框架极片可以形成较大的空间和固有的Na+扩散路径,泡沫镍的存在可以实现较为理想的电子电导率和离子扩散效率。初始容量82.6 mAh/g,大倍率6 C循环500圈后容量保持率为87.5%。这种有序地自支撑NaxMnO2纳米片阵列可以有效地缓解Mn3+反应,进而提高NaxMnO2正极材料的循环稳定性和倍率性能。
Dahn等[70]采用固相反应法合成了层状结构NaCrO2(R-3m)材料,该材料在2.0~3.6 V电压范围内具有首圈110 mAh/g的可逆容量。他们利用电弧法研究了Na0.5CrO2在350 ℃以下的热稳定性,并通过X射线衍射和电弧测试技术研究表明Na0.5CrO2分解成NaCrO2和P3层状结构的CrO2-δ以及微量的氧气,即便与同等条件下Li0.5CoO2和LiFePO4的结果相比,Na0.5CrO2也表现出更好的热稳定性[71]。该项研究成果对钠离子电池的安全性具有重大意义。Fu等[72]利用碳包覆技术使活性粒子的电化学行为得到改变,NaCrO2材料的比容量和循环稳定性有了显著提高。他们通过扫描电镜表征发现,与未进行碳包覆的NaCrO2粉末相比,碳包覆的NaCrO2具有更小的颗粒尺寸和分布均匀性,说明表面导电碳层对该正极材料的优异性能起着至关重要的作用。另外,Yabuuchi等[73]系统地研究了纳米化对NaCrO2材料储钠性能的影响。先采用机械铣削法制备了阳离子型岩盐型纳米NaCrO2原料,然后再采取岩盐相热处理法成功制备了层状O3型纳米NaCrO2。他们通过高分辨率透射电镜观察发现,次生粒子是由高结晶度的纳米级且具有丰富晶界的NaCrO2原生粒子组成。值得注意的是,不利因素O3-P3相变在这些具有丰富晶界的热处理试样中并未出现,其原因可能是不规则的晶界排列中没有出现CrO2层的滑移[74]。显然,经纳米化的NaCrO2材料展现出了更好的循环性能和结构稳定性。这些研究发现说明了纳米技术的引入可能会改变钠离子电池正极材料的设计策略,从而引领未来可充电钠离子电池技术的发展方向。
层状金属氧化物P2-NaxCoO2具有结构简单、容量大和易于合成等优点,被普遍认为是NIBs的潜在阴极材料之一[75]。最重要的是,与当前已商业化的LiCoO2相比,Na+在P2-NaxCoO2中的扩散效率(0.5~1.5×10-11 cm-2·s-1) 高于Li+在LiCoO2中扩散效率(1×10-11 cm-2·s-1)[76]。因此,针对如何提高Na+在该类材料中的扩散速率成为了研究热点。Yang等[77]首次通过“碱化和后煅烧”的方法合成了自支撑的Na0.7CoO2纳米片阵列。与传统的高温合成方法不同的是,Na0.7CoO2纳米片内足够的层间空间使该材料具有一定体积变化的可调节性,即使在充放电过程中发生了有序的相变,Na0.7CoO2纳米片阵列的整体形貌几乎没有受到任何影响。最重要的是,由于Na0.7CoO2与集流体结合紧密,巧妙地避免了活性物质的脆化和脱落,从而会提升Na+扩散速率和电导率。即使在6 C速率下循环1100圈后仍然保持51 mAh/g的可逆容量,此时容量保持率70.2%,平均库仑效率约为99.2%。Yang提出的合成方法为实现NIBs的高性能正极材料奠定了基础。日前,Pan等[78]首次将制备锂离子电池正极材料的单元前驱体合成方法(SP法)成功运用到钠离子电池正极材料上,所谓的单源前驱体(SP)法即将所涉及的金属元素以合适的比例混合在一起,由于其元素分布的均匀性和奇特的形貌,通过该方法制备的锂离子电池正极材料可以获得更为优异的电化学性能。与传统的SS方法相比,由单源前驱体NaCo(acac)3(acac=乙酰丙酮)并通过SP方法合成的P2-NaxCoO2表现出更出色的电化学性能,尤其是显著的速率性能(70 mAh/g在2000 mA/g的电流密度下),甚至优于最近报道的球形NaxCoO2(64 mAh/g在2000 mA/g的电流密度下)。这主要得益于每个元素在纳米尺度上分布的高度均匀性以及颗粒表面较高的洁净度。单元前驱体法为制备高性能的SIBs层状金属氧化物正极材料开辟了一条新的途径。
3 由二元过渡金属组成的P2结构材料
一元材料普遍存在循环寿命短、结构稳定性差和比容量低等缺点,而通过向主体材料中掺杂活性或非活性的金属离子,发挥多金属的协同作用可以明显提高一元层状材料的电化学稳定性和结构稳定性。在众多P2型过渡金属氧化物中,锰系钠离子过渡金属氧化物NayMn1-xMxO2 (x, y ≤ 1,M=Ni、Fe、Co、Ti、Cu、Mg等)得到了广泛的研究。其中,铁、锰氧化物因具有丰富的元素和良好的环保性能,展现了较为广阔的应用前景。Mn3+/Mn4+氧化还原电对可以提供较大的比容量,而Fe3+/Fe4+氧化还原电对则能提供更高的工作电位,这些都使得铁锰基氧化物成为近年来非常热门的研究课题[69,79~81]。Kalluri等[82]首次采用静电纺丝法和溶胶凝胶法分别制备了Na2/3(Fe1/2Mn1/2)O2纳米纤维和Na2/3(Fe1/2Mn1/2)O2纳米颗粒,并将其分别命名为NFMO NF和NFMO NP。通过对比两种材料的X射线衍射(XRD)图谱,这两种样品都具有良好的结晶性和P63/mmc空间群。研究发现,采用静电纺丝法制备的P2型Na2/3(Fe1/2Mn1/2)O2纳米纤维具有更优异的结构稳定性,由于纳米纤维之间互联互通,更有利于电荷转移和良好的电解质接触。该材料在SIBs中表现出良好的电化学性能,初始放电容量高达195 mAh/g,且倍率性能优异,在0.1 C的倍率下经过80次循环后,仍保持86.4%的可逆容量,显然优于同等放电条件下Na2/3(Fe1/2Mn1/2)O2纳米颗粒的可逆容量 63.1%。纳米晶粒在纳米纤维中的附着从而大大降低了纳米晶粒的自聚性,这也正是该材料具备优良的可逆性能的主要原因。这种层状结构的纳米纤维可能是SIBs的潜在正极候选材料之一。
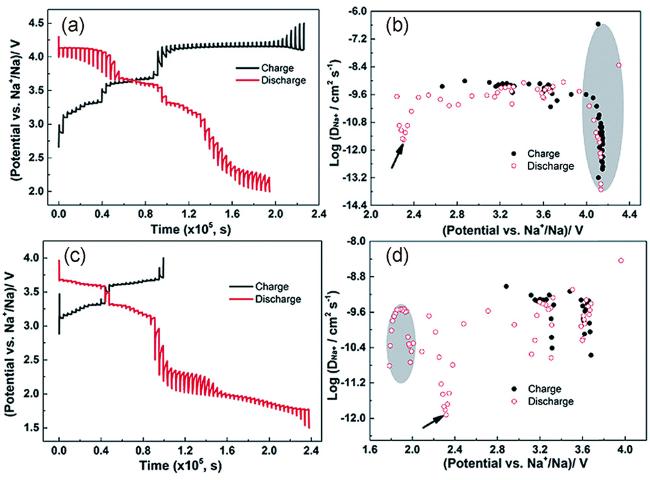
图3 (a) Na/NNMO电池首圈充放电状态的GITT曲线及(b)相应的钠离子扩散系数($D_{Na^{+}}$)在4.5 ~ 2.0 V之间;(c)Na/NNMO电池首圈充放电状态的GITT曲线以及(d) 相应的钠离子扩散系数($D_{Na^{+}}$)在2.0 ~ 1.5 V[89]Fig. 3 (a) GITT curves for the charge and discharge states of the first cycle and (b) corresponding sodium-ion diffusion coefficient ($D_{Na^{+}}$) of Na/NNMO cell cycling between 4.5 and 2.0 V;(c) GITT curves for the charge and discharge states of the first cycle and (d) corresponding $D_{Na^{+}}$ of Na/NNMO cell cycling between 2.0 and 1.5 V electrode at current rate of 6C[89] |
由于不同的相变速率和钠离子扩散速率,其电化学性能与截止电压和所用电解液高度相关。在4.0~2.0 V电压范围内以10 C循环1200圈后,电极仍有89.0 mAh/g的容量,容量保持率达到了71.2%,可见该材料具有很好的循环稳定性及结构稳定性。当截止电压设定为4.5 V或降低至1.5 V时,P2-O2相变的发生和电解质分解,或Mn4+/Mn3+离子对氧化还原反应导致容量衰减[42]。另外,Zhou等[90]通过进一步对P2-type Na2/3Ni1/3Mn2/3O2材料研究发现,该材料在2.5~4.3 V高电压范围Na+脱嵌过程中,与晶体相变相关的过渡金属氧化物层剥落是导致容量衰减的主要原因,为解决上述问题,他们利用Al2O3涂层技术的优势较为明显地改善了Al2O3-Na2/3[Ni1/3Mn2/3]O2材料的循环稳定性。而未进行表面修饰的Na2/3Ni1/3Mn2/3O2样品在电压为2.5~4.3 V内的初始放电容量为164 mAh/g,循环300圈后容量衰减至44 mAh/g,容量保持率仅为26.8%。相比之下,经过Al2O3包覆的Na2/3[Ni1/3Mn2/3]O2具有类似的初始容量,经300次循环后容量保留率提高到了73.2%。以上研究结果表明,Al2O3表面涂层这一策略不仅能够有效抑制高电压下的不良副反应和金属氧化物层的剥落,还能对电极材料的结构起到更好的保护作用[91,92]。
Lim等[93]采用传统的固相法合成了P2型Na0.6Ca0.07CoO2正极材料,由于位于CoO6层的Ca2+增强了静电吸引力,导致单位晶胞体积不断收缩,使得Na0.6Ca0.07CoO2具有更稳定的容量保持能力。另外,晶格中的Ca2+减轻了Na+的空位有序性对电化学性能带来的负面影响,使得该材料在循环性能(经循环60周后,比容量由105 mAh/g减少至101 mAh/g)和速率方面都大幅地提升。
通常情况下,含镍或钴元素的层状电极材料具有较高的存储容量、速率快和循环稳定性好等优点[94,95,96]。Manikandan等[97]通过溶胶凝胶法制备了P2层状正极材料Na0.5Ni0.25Mn0.75O2,在放电区间1.5~4.4 V具有210 mAh/g的放电容量,以0.1 C循环50周仍有80%的可逆容量,循环稳定性表现不错,值得关注的是该材料在高电压4.1 V充放电曲线极为平滑且对应80 mAh/g的比容量,优于报道过的大多数钠离子电池正极材料。Dai等[98]用聚乙烯吡咯烷酮(PVP)凝胶燃烧法合成了P2 Na2/3Ni1/3Mn2/3O2材料并研究了其电化学性能,该合成方法与其他聚合物热解法相比具有显著优点,活性物质在原子水平上的分布会更加均匀,提高了Na2/3Ni1/3Mn2/3O2的结晶性、纯度和均匀性。该材料在2.3~4.1 V可给出87.6 mAh/g的可逆容量,经5 C循环650周后具有89%的容量保持率,循环稳定性非常出色。 此外,与硬碳为对电极组合的全电池理论能量密度达210 Wh/kg,具有很高的实际应用价值。Carlier等[99]研究了P2型Na2/3Co2/3Mn1/3O2材料的结构和电化学性质,该材料中低自旋态的Co3+和Mn4+是稳定的,并随机分布在过渡金属层内,钠离子位于Naf和Nae层之间,且优先占据Nae位点,该材料在1.5~4.0 V展现出良好的可逆性,可实现>0.5Na的嵌脱反应。Delmas等[100]采用原位和原位X射线吸收光谱法重新研究了Na2/3Co2/3Mn1/3O2中钠离子的插入,结合密度泛函理论(DFT)和X射线吸收光谱(XAS)数据,阐明了在钠离子插入过程中O-K边界的电子跃迁和电荷补偿机制,发现Co3+/Co2+和Mn4+/Mn3+的氧化还原电对同时参与了电荷补偿过程。
为了进一步提高此类材料的倍率性能和循环性能,Zhou等[101]通过传统的溶胶-凝胶法合成了P2-Na0.67Co0.5Mn0.5O2,该材料0.1 C可给出147 mAh/g的高可逆容量,以1 C循环100周后容量保持率接近100%,循环稳定性十分优异。同时,该材料在30 C的电流密度下(图4),表现出非常高的稳定性。同时,他们结合电化学循环原位XRD检测材料的结构变化发现(图5),主要原因是在钠离子脱嵌过程中只存在单相固溶反应,没有不可逆的相变。但是,较低的平均工作电压仍然是Co-Mn基层状氧化物材料存在的致命问题。Manikandan等[102]通过混合氢氧碳酸盐方法得到了高压层状Na0.5Co0.5Mn0.5O2正极材料,这一化合物在1.5~4.4 V可实现Co3+/Co4+和Mn4+/Mn3+的氧化还原,分别对应4.19 V和2.3 V高压氧化峰。首周充放电容量53/144 mAh/g以及较好的倍率性能。Hemalatha等[103]采用尿素-甘氨酸助燃法制备了一系列钴取代的P2-Na0.67MnxCo1 -xO2(x=0.25, 0.5, 0.75)的化合物,发现钴取代可以降低晶格参数a和c,导致MO6八面体收缩,同时增大d层间间距,这为钠离子扩散提供更大的空间。同时,结合XPS结果表明,Na0.67MnO2中的等价钴取代部分/完全取代了引起Jahn Teller的活性Mn3+,形成了结构稳定性较好的低自旋配合物。这使材料的倍率性能、循环性能和结构稳定性均有大幅提升。其中,Na0.67Mn0.75Co0.25O2(x=0.75)的材料具有167 mAh/g的放电容量,循环100周后仍有126 mAh/g的可逆容量,相较于另两个材料,表现出较高的容量。同时,他们对材料中的钴含量对反应过程中的钠离子扩散系数($D_{Na^{+}}$)进行了详细分析, $D_{Na^{+}}$值会随着钴含量的增加而增加,依次为MC25 > MC50 > MC75,然而容量保持率却呈相反趋势。

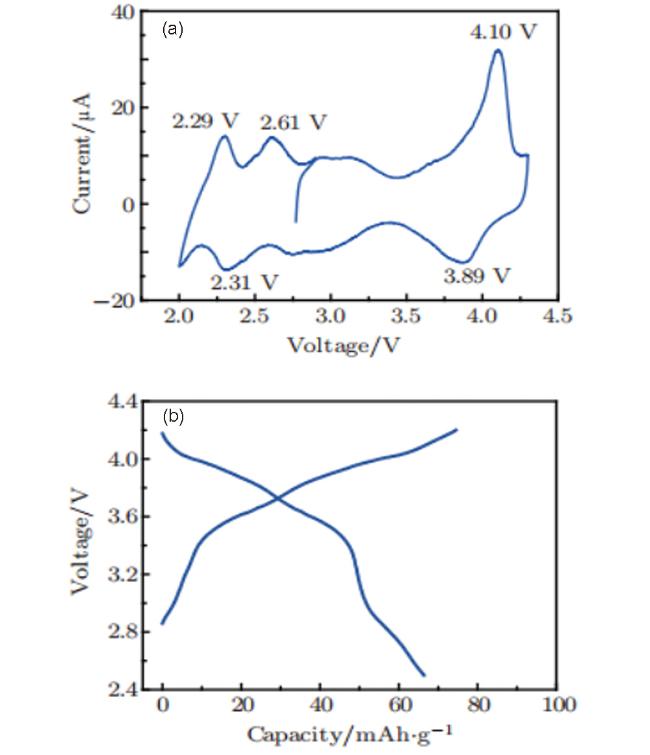
考虑到镍、钴元素不仅有毒,而且价格相对昂贵,使用这些元素势必会提高电池的成本,不利于大规模的储能应用。为此,Chen等[104]首次发现廉价无毒的Cu2+/Cu3+氧化还原电对具有电化学活性,他们采用固相法制备了P2型层状材料Na0.68Cu0.34Mn0.66O2,图6a为该材料在2.0~4.3 V的循环伏安曲线,其中位于4.1 V和3.9 V的一对氧化还原峰分别对应Cu3+/Cu2+的氧化还原峰对。图6b表明该材料具有74.5 mAh/g初始比容量,对应于0.3 mol Na+从Na0.68Cu0.34Mn0.66O2中脱出且极化很小,由于这里Mn已经是+4了,此时Cu2+氧化为Cu3+。虽然该材料的比容量略低,但这一重大发现无疑对探索低成本和高比容量的钠离子电池材料具有重大的意义。
4 由三元过渡金属组成的P2结构材料
Man等[105]采用陶瓷法合成了高性能的富锰P2型Na2/3Mn0.8Fe0.1Ti0.1O2材料,该材料的第二次充放电比容量为146.57/144.16 mAh/g,以C/10的放电速率在电压范围为4.0~2.0 V内,经循环50圈后仍保留95.09%的放电容量。当1 C的放电倍率经过2~300圈循环后时,可逆比容量达到99.40 mAh/g,容量保持率达到87.70%。微量过渡金属Ti的掺杂使得材料的结构稳定性和电化学性能大幅高。Zhou等[106]通过向Na0.67Fe0.5Mn0.5O2中掺杂少量的Co元素,得到了在速率性能和容量保留率方面都有所提高的Na0.67Fe0.3Mn0.3Co0.4O2材料。研究发现Co的引入可提高结构中Na+的扩散速率,与Na0.67Fe0.5Mn0.5O2相比,Na0.67Fe0.3Mn0.3Co0.4O2正极材料表现出了优异的容量保持能力(以1 C放电速率循环100次后,仍有85.5%的初始容量)以及较好的倍率性能(以0.2 C时为136.7 mAh/g,5 C时为81.1 mAh/g)。另外,该研究也揭示了过渡金属Co的掺杂有效地提高材料电子导电性的同时在一定程度上缓解了电极极化[107]。
Na2/3Ni1/3Mn2/3O2(NNMO)是一种典型P2型层状结构材料,该材料虽然具有较高的理论容量和工作电压,但由于充电至4.2 V时,P2-O3相变的存在致使体积发生变化,使得循环稳定性较差[108]。为了优化此类材料的电化学性能,Komaba等[109]研究发现通过Ti取代NNMO中部分Mn得到的Na2/3Ni1/3Mn1/2Ti1/6O2具有127 mAh/g的可逆容量,在2.5~4.5 V电压范围可得到3.7 V的平均放电电压,循环性能也十分优越。另外,该材料与硬碳为对电极组装的全电池具有300 Wh/kg以上的能量密度,约为石墨//LiCoO2系统能量密度的80%。Zhang等[110]将具有电化学活性的元素Fe3+引入NNMO晶格中,合成了具有可逆性强和稳定性高的晶体结构P2型Na2/3Ni1/3Mn7/12Fe1/12O2材料。Fe3+的引入通过降低Na+的有序性使得该材料不仅具有优异的倍率性能和较长的循环稳定性(图7),而且P2~O3的相变也得到了抑制,既使在-25 ℃的低温下也没有出现容量大幅衰减的迹象。通过对比图8a和8b表明,在充满电荷的1/12-NNMF中(002)晶面没有出现明显的偏移和分裂,说明不存在OP4结构和“Z”相,即说明在1/12-NNMF电极中P2-O2不可逆相变得到抑制。适量的Fe取代Mn可以有效抑制相变,增强结构稳定性,促进Na+扩散,在不需要牺牲太多容量的情况下,还能获得更好的电化学性能。与此同时,Xu等[45]曾尝试将Co3+掺杂到Na2/3Ni1/3Mn2/3O2(NNMO)中,获得了较为理想的588 Wh/kg能量密度的Na2/3[Ni0.3Co0.1Mn0.6]O2材料,其能量密度可与商业化的LiCoO2和Li[NixCoyMn1- x-y]O2(x ≤0.5)正极相比,并高于应用于LIB系统中的橄榄石LiFePO4和尖晶石LiMn2O4正极[53]。在2.0~4.3 V电压范围以0.1 C经过50次循环,仍然有128 mAh/g的高可逆容量,容量保持率为79.2%。值得注意的是,Co3+的移入不但没有降低能量密度,而且还抑制了P2-O2相变。该材料与硬碳作为对电极组成的全电池具有150.6 mAh/g的有效容量,在0.1 C倍率下的平均电压为3.5 V,以0.5 C循环100圈循环后的可逆容量为86 mAh/g,对应的容量保持率为70.2%,在10 C大倍率下仍有92 mAh/g,展现出诱人的实际应用前景。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Wang等[111]在过渡金属晶格中引入Cu2+也是防止材料在Na+脱嵌后发生相变的一种有效策略,具有电化学活性的Cu2+不仅可以抑制P2型层状材料在高电位存在的P2-O2相变和降低Na+空位有序性,而且由于Cu2+/Cu3+具有较高的氧化还原电位,还有助于提升可充电容量。他们通过简单的溶胶-凝胶法合成了结构稳定性和储钠性能优异的P2结构层状复合材料Na0.67Ni0.1Cu0.2Mn0.7O2,在Na0.67Ni0.3Mn0.7O2中出现的结构退化问题也得到了解决。但是,由于铜的相对原子质量比镍的相对原子质量大,导致该Na0.67Ni0.1Cu0.2Mn0.7O2材料仅有114.7 mAh/g初始放电容量,低于未掺杂Cu2+的Na0.67Ni0.3Mn0.7O2的151 mAh/g。
与Mn半径类似且具有低成本的电化学惰性Al元素对稳定层状结构及提高电极性能也有一定的影响。Hasa等[43]采用共沉淀法制备的Na0.6Ni0.22Al0.11Mn0.66O2(NAM)材料表现出十分优异的循环性能,在较宽的电压区间1.5~4.6 V以0.1 C和0.5 C循环时分别具有 250 mAh/g 和150 mAh/g 的可逆容量。此外,当电流从5 C回到0.1 C时,该电池可以恢复约91%的初始容量,这表明Al元素的掺杂可以有效地增强材料在高压下P2-O2相变的可逆性。
5 P2结构材料存在的问题及优化改性
5.1 P2结构材料存在的问题
目前,钠离子电池P2型正极材料仍存在一些问题亟待解决。由于Na+离子半径大,Na+在迁移过程中存在着很明显的动力学障碍,以及在脱嵌过程中对材料结构造成较大的影响,例如体积膨胀、结构坍塌等,导致钠离子电池存在倍率性能较差和循环衰减较快的问题,尤其是电位处于截止电压时,可逆容量更低。大多数P2型材料因存在不可逆的相变使得初始库仑效率较低和不够理想的工作电压,从而限制其走向实际应用的发展。例如,P2结构的Na2/3Ni1/3Mn2/3O2(P2-NNM)材料,Wang等[114]采用先进的透射电镜研究发现,定性证明该材料在高充电截止电压下性能衰减较快的原因是由反复的P2-O2相变引起的晶内裂纹来分解正极材料的原生颗粒,而不是由表面退化引起的。
5.2 掺杂改性
Zhou等[115]以溶胶凝胶法在800 ℃制备了P2/P3复合层状材料Na0.7Li0.06Mg0.06Ni0.22Mn0.67O2,并详细研究了合成温度、材料结构以及电化学性能之间的关系。通过原位XRD发现,复合材料中P3(占46.7%)和P2(占53.3%)相分别形成于670 ℃和750 ℃,而纯的P3和P2相分别形成于750 ℃以下和830 ℃以上。该材料兼具P2、P3两种层状材料的结构优势,可为Na+提供较大扩散通道以及具有高度可逆的P2/P3-OP4/P3相变。结合X射线光电子能谱(XPS)发现在充放电区间2.0~4.4 V Ni2+完全氧化为Ni4+,对应119 mAh/g的可逆比容量以及94.8%的初始库仑效率。与单相P型层状材料相比,P2/P3复合层状材料具有出色的循环稳定性和良好的动力学性能,与硬碳为负极组合的全电池理论能量密度可达218 Wh/kg,循环50圈后仍有97.2%的容量保持率,即使在大倍率5 C的情况下依然有85.55%容量。
基于高容量、低成本的层状P2结构的Na0.67Fe0.5Mn0.5O2材料因其组成成分廉价、易得而被认为是一种极具应用潜力的钠离子电池正极材料。然而,该材料在倍率性和循环稳定性方面表现很差。为解决上述问题,Zhou等[116]向P2 Na0.67Fe0.5Mn0.5O2中引入适量的Mg2+并系统研究了Mg2+对正极材料形貌、结构以及电化学性能的影响。研究表明,经过优化处理后可以得到P2/O3两相共存的Na0.67Fe0.425Mn0.425Mg0.15O2材料,在P2和O3两相的协同作用下,该材料的循环稳定性和倍率性能都有效地得到大幅提高,该材料具有98.1 mAh/g 初始放电容量,在1 C倍率下经过50次循环后,容量保持率具有惊人的95.7%。经过100次循环后,仍能保持87.7%的容量。
目前,P2型Na-Mn-Ni-O正极材料引起众多学者的深入研究。普遍采用阳离子掺杂进行晶格调控和抑制不可逆的P2-O2相变,例如使用元素Zn2+、Al3+、Cu2+或者Ti4+代替部分Mn[109, 117~119],使材料的电化学性能、循环寿命等得到了实质性的提高。然而对于具有高工作电位和高可逆容量的活性Ni2+/Ni3+氧化还原电对尚未得到广泛研究。近日,Chen等[120]提出了使用阴离子F-掺杂策略可以进一步提高Ni2+的活性。研究表明,掺杂的氟离子位于氧位上,与钠可形成2个稳定的化学键F—Na,可以有效地防止因Na+过量脱嵌而引起P2相结构的坍塌,提高材料的循环稳定性,在2 C的倍率下循环960次后容量保留率为75.0%。此外,该研究还发现经过F-掺后的材料的比容量和Na+扩散速率都有明显的提高。掺杂F-的材料经优化处理后得到的晶格参数(a=b=2.8856, c=11.276)与原始材料(a=b=2.8826, c=11.247)相比,晶格参数c的略有增加可改善Na+在插入/提取过程中的动力学。
一般情况下,不同类型的过渡金属的费米能级相近时就会导致Na+/空位有序性发生,从而会影响到位于过渡金属层间的Na+离子扩散速率。因此,考虑到Ni2+和Mg2+的价电子相同,但费米能级有明显差异,在TMO2层中用Mg2+代替Ni2+可以有效地抑制TMs有序性和电荷有序性、Na+/空位有序性等缺点[121,122,123]。Tie等[124]设计开发出一种由Ni2+ 和Mg2+共掺杂的富Mn Na0.67Ni0.1Mn0.8Mg0.1O2钠离子电池正极材料。不仅克服了Mn3+的Jahn-Teller效应,而且由于Ni2+的存在电压平台也随之提高。通过电化学性能测试数据表明,样品在0.1 C时的放电比容量为193 mAh/g,循环100次后的容量保持率为74.7%,在高倍率8 C时的比容量为70 mAh/g。通过深入研究动力学表明,Na0.67Ni0.1Mn0.8Mg0.1O2的扩散系数(D)较与未进行Ni和Mg掺杂的样品有较明显的提高。Na+迁移率的提高可能是Na+/空位有序性的减少和过渡金属层层间距离增大的原因。此外,Ni和Mg的共掺杂对材料层状结构的稳定性也有突出的贡献。
5.3 表面修饰改性
虽然不少国内外的研究小组对钠离子电池正极层状材料做了大量的研究,通过掺杂阳离子或阴离子、不同相之间的混合等,从而大幅降低储能系统的成本,提高储能系统的长期可靠性和安全稳定性。由于钠离子半径比锂离子半径大的多,在宿主材料中Na+的脱嵌要比Li+困难得多,不但扩散速率不如锂离子电池,而且在循环过程中还面临着结构变化较大的问题,导致容量衰退较快和循环寿命较短的缺点。除此之外,其较高的空气敏感性对材料的电化学性能也会带来负面作用。为此,研究者们通过对材料表面进行修饰来阻止循环过程中活性物质与电解液的直接接触,避免正极材料与电解液发生副反应,使得电极材料的离子扩散能力、电子导电性和结构稳定性都有较大的提升。例如对材料表面采用碳包覆的策略来提高电极材料电化学性能,可是碳的机械性能较差,导致碳涂层层状氧化物对材料循环稳定性的提高非常有限[125]。为此,Kaliyappan等[35]曾尝试过对材料表面进行 Al2O3包覆来改善材料的循环稳定性,但是他们发现较厚的Al2O3涂层严重降低了活性材料的利用率,增加了循环时的电荷转移电阻,导致倍率性能很差。
近日,Chu等[126]同样利用原子层沉积技术在Na2/3Mn1/2Fe1/4Co1/4O2电极上制备了纳米级厚度的Al2O3涂层。与原材料相比,超薄的Al2O3涂层并没有阻碍钠离子的插入/提取和降低电极的比容量,经涂层后的电极材料对空气中的H2O和CO2敏感度也有明显降低,可以有效地缓解这种缺陷。根据CV和EIS表征发现,Al2O3涂层的使用在抑制液体电解质与活性材料界面处在高电位下发生的副反应发挥着巨大的作用,即使在4.5 V的高截止电压下仍能实现217.9 mAh/g的容量,以0.5 C在1.5~4.5 V充放电循环100次以上,容量保留率高达84.8%,表明电化学稳定性有一定的改善。为了进一步提升P2-Na0.67Co0.25Mn0.75O2材料的循环稳定性和动力学性质,Tang等[127]通过简单的固相法合成了Na0.67Co0.25Mn0.75O2氧化物,并用CeO2对其表面进行修饰,该材料在2.0~4.0 V之间有着135.93 mAh/g 的放电容量,以20 mAh/g电流密度循环50和100圈后的容量保持率分别为91.96%和83.38%,容量保持率较之前有显著的提高。上述的Al2O3涂层技术、碳包覆和CeO2包覆在锂离子电池和钠离子电池领域都已做了大量的研究,但是,关于金属磷酸盐改性钠离子电池层状氧化物材料的研究工作却鲜有报道。Wang等[128]以AlPO4和Mg3(PO4)2为涂层材料,分别采用共沉淀法和固相烧结法制备了Na0.65Mn0.75Ni0.25O2/AlPO4和Na0.65Mn0.75Ni0.25O2/Mg3(PO4)2两种样品。据XRD图谱分析,与AlPO4或Mg3(PO4)2相对应的衍射图谱并没有出现,表明少量的金属磷酸盐涂层对原始样品(Na0.65Mn0.75Ni0.25O2 )的晶体结构并不会产生影响,并具有较高的可逆容量和充放电平台。他们从微观结构发现,通过比较原始材料和经金属磷酸盐涂层的材料在空气中暴露后的XRD图谱发现,(100)晶格面在金属磷酸盐包覆后生长受到抑制是活性物质材料能不受到空气中H2O或O2侵蚀的主要原因,三种材料在0.2 C循环100圈时,容量保持率分别为57.1%、80.1%和78.7%,说明经磷酸盐包覆的材料循环性能好,此外,由于表面Na3PO4和AlPO4的存在,对提升电导率也有效果,从而速率性能也得到改进。综上所述,表面修饰技术是提高层状电极材料循环寿命的重要途径之一,也是未来的研究重点,从而加速推进钠离子电池技术商业化的进程。
6 阴离子氧化还原提高首次充电比容量
近年来,含有阴离子氧化还原电对的正极材料已成为十分热门的研究领域,因为借助这一概念可以获得超过阳离子氧化还原限制的容量。一些研究表明,通过激活P2结构材料氧离子的氧化还原(Oxygen redox,简称OR),可以有效提高首次充电容量,在一定程度上弥补首次充电容量的短板。Yabuuchi等[84]制备的Na2/3[Mg0.28Mn0.72]O2,得益于OR[131],在10 mA/g电流下首次充电至4.4 V容量达150 mAh/g。Dai等[132]进一步制备了Mn理论价态为+4价的Na2/3Mg1/3Mn2/3O2,在0.1 C倍率下充电至4.5 V,首次充电容量可达162 mAh/g。虽然初始材料Mn非常接近+4价(Mn4+占95%),然而Mg离子的加入,激活了OR。研究通过Mn-L mRIXS-iPFY精确测定了充放电过程中体相Mn的价态演变,发现在充电过程中及放电的前半段Mn保持+4价,并进一步通过超高效率共振非弹性X射线散射全谱图(mRIXS)对Na2/3Mg1/3Mn2/3O2中的OR可逆性和循环保持率进行了精确可靠的定量分析,发现其中的OR反应高度可逆且循环保持良好,这也表明了在廉价的第三周期过渡金属氧化物材料中实现高度可逆且稳定循环的OR反应是可行的。
除了Mg之外,Li的加入也可以激活OR。Rong等[39]制备了P2结构的Na0.72[Li0.24Mn0.76]O2,首次充电容量可达210 mAh/g。通过Mn-K XANES研究了充放电过程中体相Mn价态演变,结合O-K XAS,确证了其中OR的存在。他们发现材料在如此大量的Na嵌入脱出的情况下仍能保持稳定的原因是阴离子氧化还原能抑制P2/O2相转变,从而使材料具有低应变特性。这打破了过去对阴离子氧化还原参与电化学循环对循环稳定性不利的固有认识,为开发长寿命高能量密度的钠离子电池正极材料奠定了基础。
然而,需要看到的是,阴离子氧化还原参与的正极材料,还存在着诸如充放电电压迟滞(充放电曲线不可逆,电压平台相差较大)、放电平台随循环衰减和倍率循环性能较差等问题。这就需要人们对其中的阴离子氧化还原进行有针对性的调控,而这一调控的基础,就在于界定不同充放电阶段不同的氧化还原电对。
7 结论与展望
虽然钠在地壳中丰度很高,钠化合物原料价格很低,然而,碱金属离子电池中碱金属原材料成本占比很低,而钠离子电池的能量密度尚不及锂离子电池。钠离子电池如果要走向实用,就必须提高能量密度,降低每瓦时成本。因此,发展高能量密度(既包括质量密度,也包括体积密度)的材料是钠离子电池正极材料的最优先发展方向。
P2结构正极材料相对其他种类的钠离子电池正极材料具有体积和质量能量密度的优势,近年来发展迅速。从一元材料发展到二元、三元乃至多元复合过渡金属氧化物,并通过少量元素掺杂和表面修饰对材料性能进行了进一步优化,已经展现出实用化前景。
目前该类材料仍待解决的核心问题为提高首次充电比容量。借助结构中氧的氧化还原反应,是解决这一问题的重要方向,目前已经看到了一些曙光。然而,阴离子氧化还原机理目前仍不清晰,以及由此带来的电压迟滞、放电平台随循环衰减、倍率循环性能较差等问题还需要进行大量的研究。这需要人们对氧的氧化[100]还原过程进行热力学和动力学性质的调控,从而提高材料的性能。借助一些定量表征手段(如mRIXS),可以区分充放电过程中过渡金属和氧的不同反应,这为专门针对不同氧化还原阶段的研究和调控打下了基础,未来的研究可以分别找到改进不同氧化还原阶段的较优手段,进一步综合这些手段,就有望进一步提高这一材料的能量密度、能量效率、倍率性能和循环性能。
此外,该材料的倍率性能、循环寿命、存储性能、高低温性能、安全性、成本仍需进一步优化和深入研究。进一步优化材料组成、制备条件和改性方法,采用储量丰富、价格低廉、安全环保的原材料,以进一步提高性能、降低成本和保护环境也是该材料的重要研究方向。