



Contents
1 Introduction
2 Coprecipitation method
3 Successive reduction method
4 Wet-impregnation method
5 Other methods
6 Conclusion and outlook
1 引言
表1 常用的单原子催化剂制备方法Table 1 Commonly used preparation methods for single atom catalysts |
| Preparation method | Catalyst | Carrier | Reaction catalyzed | Advantages | Disadvantages | Ref |
|---|---|---|---|---|---|---|
| Coprecipitation | Pt | FeO x | CO oxidation | even distribution of active single atoms on carrier | catalytic activity susceptible to many factors and low load | 17 |
| Ir | FeO x | water gas conversion | 8 | |||
| Ag | Hollandite-type MnO2 | Hydrogenation of glyoxylate | 35 | |||
| 34 | ||||||
| PtAu | MCNTs | formic acid oxidation | ||||
| Successive reduction method | Au | Pd nanocluster | glucose oxidation | excellent catalytic activity and long shelf life | complicated preparation and hard-to-control structure | 30 |
| Au | IrPd nanocluster | glucose oxidation | 36 | |||
| Au | Pd nanocluster | glucose oxidation | 37 | |||
| Pd1 | Au33or Au43 | benzyl alcohol oxidation | 31 | |||
| NiCu | SiO2 | ethanol dehydrogenation | 38 | |||
| Wet-impregmation method | Pt | Fe-N-C | ORR | facile process and no need for specific appaaratus | Low load | 39 |
| Rh | ZnO | Hydroformylation of olefins | 40 | |||
| Pt | Sb-doped tin oxide | formic acid oxidation | 41 | |||
| CoN | graphene | Cathode catalyst of Zn air battery | 42 | |||
| Pt | θ-Al2O3 | CO oxidation | 43 | |||
| Ni | graphene | Electrocatalytic hydrogen evolution | 44 | |||
| Au | TiO2 | water gas conversion | 45 |
2 共沉淀法制备单原子催化剂
Zhang等[17]以7.59×10-2 mol·L-1的H2PtCl6溶液和1.0 mol·L-1的Fe(NO3)3溶液为催化剂前驱体,采用共沉淀法制备了Pt1/FeO x 单原子催化剂。他们在50 ℃下调节溶液的pH=8得到前驱体沉淀,先过滤干燥后,再经H2还原得到负载量为0.17 wt%的Pt1/FeO x 单原子催化剂。从催化剂的高角环形暗场像-透射电子显微镜(HAADF-STEM)图像(图1)和X射线吸收近边(XANES)谱的结果中可知:催化剂中Pt原子将电子转移给FeO x ,使得Pt带正电。另外,由于Pt原子与载体FeO x 中的O原子存在着强相互作用,单原子Pt位于FeO x 表面的缺陷处,因而能够稳定地负载在FeO x 表面,并具有优异的催化性能。密度泛函理论(DFT)的计算结果表明:经H2处理后,Pt单原子周围的FeO x 被部分还原,产生了O空位,在该空位处可吸附一个O2分子;Pt单原子使O2分子活化,降低其吸附能。与Pt纳米团簇相比,Pt单原子对CO分子的吸附能更低,使其与一个O原子结合生成一个CO2分子。第二个CO分子吸附后与余下的一个O原子结合生成另一个CO2分子,形成一个完整的循环。之后在该位置处又形成一个新的O空位,持续重复以上过程,每次循环可生成2个CO2分子。Pt1/FeO x 单原子催化剂在催化氧化CO反应中表现出优异的活性,且转化率接近100%;负载量为0.17 wt%的Pt1/FeO x 单原子催化剂的催化活性值是负载量为2.5 wt%和4.4 wt%的2~3倍。

Zhang等[8]以H2IrCl6为催化剂前驱体,以FeO x 为载体,采用共沉淀法制备了负载量为0.01 wt%的Ir1/FeO x 单原子催化剂。HAADF-STEM结果表明:Ir1/FeO x 单原子催化剂中仅存在单原子Ir;而负载量为2.40 wt%时,催化剂中除了Ir单原子外,还存在尺寸小于2 nm的Ir纳米团簇。不同负载量的Ir/FeO x 单原子催化剂的反应活化能接近,说明这些催化剂具有相同的催化活性位点。在催化反应中,Ir原子起着决定性的作用。氢气程序升温还原(TPR)和程序升温表面反应(TPSR)的研究结果表明:单原子Ir的存在降低了FeO x 的还原温度,Ir/FeO x 的催化活性高于FeO x ,且优先产生H2而非CO2;他们推测Ir1/FeO x 单原子催化剂的催化机理如下:单原子Ir降低了FeO x 的还原温度促进其还原,并在FeO x 的表面产生大量的氧空位,H2O分子附着在氧空位上并分解产生H2和O*;CO与O*结合生成CO2。Ir单原子锚定于还原后的FeO x 表面,单原子Ir和FeO x 之间的协同作用使该单原子催化剂具有高的催化活性。当负载量为0.01 wt%时,Ir1/FeO x 单原子催化剂具有最高的水煤气催化反应活性,其催化活性是传统的Ir/FeO x 载体型催化剂的7~19倍,也比其他的Ir纳米团簇催化剂高出了2~3个数量级。由于单原子催化剂负载量的增多会导致纳米团簇的出现,进而降低催化剂的催化活性。因此,采用这种方法制备的单原子催化剂的负载量较低,载体的利用率较低,提高了催化剂的成本。
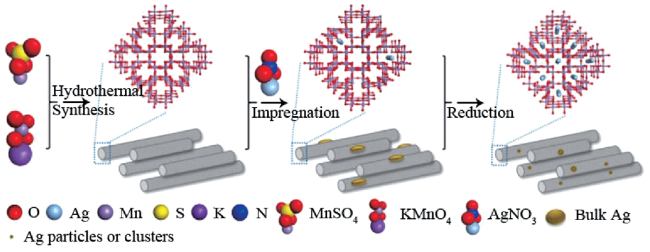
为了解决单原子催化剂低负载量的问题,Ding等[35]采用具有特殊孔道结构的MnO2为载体来提高贵金属的负载量。他们以MnSO4及KMnO4为原料,AgNO3为催化剂前驱体,采用共沉淀法制备了Ag/Hollandite型MnO2单原子催化剂。先通过水热法制备Hollandite型MnO2,随后将所制备的[Ag(NH3)2]OH溶液和H2O2溶液加入到MnO2悬浮液中,干燥后得到负载量约为28.8 wt%的Ag/Hollandite型MnO2单原子催化剂。结果表明:Hollandite型MnO2的截面上存在着0.47×0.47 nm2的独特方形隧道结构,这些隧道结构的基本单元由8个氧原子构成,该结构和氧原子的存在有利于Ag原子在隧道中的稳定存在,其形成过程示意图如图2所示。XPS结果表明:还原后的Ag/Hollandite型MnO2单原子催化剂中仅存在单质Ag而非AgO。由H2-程序升温脱附(TPD)曲线可知,Hollandite型MnO2可通过化学吸附来吸附H2,而Ag纳米团簇不会;他们认为较高的催化活性可能是由于Ag原子进入Hollandite型MnO2晶格隧道中,导致Mn周围电子密度的降低。晶格隧道中的单原子Ag与Hollandite型MnO2之间存在着的强相互作用,使催化剂在低温下即可吸附表面的H2。单原子催化剂对乙二醇脱水的活化能为296.78 kJ/mol,可以在低温下高选择性地生成乙醇酸乙酯,并进而在高温下生成乙醛缩二乙醇。
傅里叶漫反射红外光谱(DRIFTS)的结果表明:Hollandite型MnO2表现出非常弱的乙醇吸附,而Ag/Hollandite型MnO2单原子催化剂则表现出强的吸附峰,表明原子状态Ag的存在对乙醇的吸附起着重要作用,并且随着温度的升高,该吸附峰的峰强也增加。相比之下,Hollandite型MnO2几乎没有表现出峰。
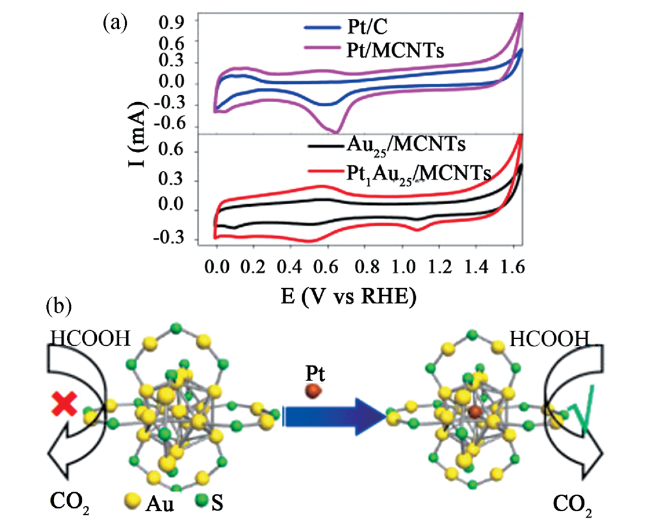
Lu等[34]以HAuCl4及H2PtCl6为催化剂前驱体,四辛基溴化铵(TOAB)为表面活性剂,多壁碳纳米管(MCNTs)为载体,采用共沉淀法制备了Pt1Au24/多壁碳纳米管单原子催化剂。他们先将HAuCl4·4H2O、H2PtCl6·6H2O和TOAB均匀混合,然后向其中加入四氢呋喃(THF)溶剂,搅拌均匀后先缓慢加入1-十二烷硫醇(1-C12H25SH),再加入NaBH4溶液还原并收集沉淀物,经洗涤后得到Pt1Au24(SC12H25)18团簇,最后将其吸附在经酸化处理的多壁碳纳米管上制备Pt1Au24/多壁碳纳米管单原子催化剂。催化剂的CV结果(图3a)表明:Pt1Au24团簇显示出与Au25团簇相似的分布,但与Pt/C和Pt/多壁碳纳米管催化剂的CV曲线有明显的不同,说明Pt1Au24团簇表面没有Pt原子,Pt原子应位于(Au24)簇的中心,如图3b所示。密度泛函理论(DFT)的计算结果表明:甲酸(HCOOH)和羧基(-COOH)中间体在Pt1Au24纳米团簇上具有更高的结合能,使该催化剂具有较高的催化甲酸氧化活性,是Pt纳米团簇的12倍,商业Pt/C催化剂的34倍。共沉淀法很难制备高负载量的单原子催化剂,这是由于金属氧化物载体的表面物相分布不均一且配位不饱和度低,导致单原子催化剂易团聚,致使单原子催化剂活性降低。且利用该方法制备单原子催化剂时,需要考虑的影响因素较多,如前驱体溶液的滴加顺序和速度、液滴的大小、温度及pH值等,不利于催化剂的高效制备及应用。
3 逐步还原法制备单原子催化剂
逐步还原法需要先制备一种纳米团簇,随后再采用置换反应还原、连续还原或电化学沉积等方法在该纳米团簇表面生成另一种金属原子,进而制得金属纳米团簇负载的单原子催化剂。
2011年,本课题组[46]先采用醇还原法制备PVP保护的Pd纳米团簇,再采用置换反应的方法制备了皇冠-明珠结构(Crown-Jewel) Au/Pd单原子催化剂。在Au/Pd单原子催化剂的制备中,Pd纳米团簇表面顶点位置的原子与Au原子发生置换反应,其合成示意图如图4所示。结果表明:在置换反应中Au原子取代了位于顶点位置的Pd原子,且每生成2个Au单原子要消耗3个Pd原子。电子能量损失谱(EELS)的结果表明:有部分Au原子位于Pd纳米团簇表面的顶点位置;实验及DFT计算结果证实了Au/Pd 单原子催化剂的高活性来源于Au和Pd原子之间的电荷转移效应,团簇中的Pd原子向Au原子转移电荷,使得Au原子显电负性;在催化葡萄糖氧化反应的过程中,Au原子又将电荷转移给O2,活化了O2分子,从而使催化反应高效地进行。(Crown-Jewel) Au/Pd单原子催化剂催化葡萄糖氧化反应的催化活性值高达195 000 molglucose/h·mol-Au,是Au或Pd单金属纳米团簇的20~30倍,是Au/Pd双金属纳米团簇的8~10倍。

(Crown-Jewel) Au/Pt单原子催化剂[1]也可采用类似的方法加以合成,研究结果表明:Au单原子位于Pt纳米团簇的顶点位置,该催化剂催化葡萄糖氧化的活性为134 700 molglucose/h·mol-Au,是相同粒径的Au纳米团簇的15倍,是相同粒径的Au/Pt 双金属纳米团簇的4倍。
在此基础上,本课题组[36]还以HAuCl4·4H2O、IrCl3及PdCl2为催化剂前驱体,以PVP为保护剂,先采用醇还原的方法制备了IrPd双金属纳米团簇,再采用置换反应法制备了PVP保护的三金属的Jewel-Structured (IrPd)/Au单原子催化剂;实验及密度泛函理论(DFT)的计算结果表明:三种金属之间的电荷转移协同效应使该单原子催化剂催化葡萄糖氧化的催化活性值高达343 190 molglucose/h·mol-Au,是相同粒径Au、Pd或Ir单金属纳米团簇的40倍,是Au/Pd双金属纳米团簇的14倍。
连续还原法也可以用来制备金属纳米团簇负载的单原子催化剂。本课题组[37]先采用醇还原法制备出PVP保护的Pd纳米团簇溶液,随后将PVP/HAuCl4溶液快速加入到PVP/L-抗坏血酸Pd纳米团簇溶胶中,再将混合液体在95 ℃的水浴中加热15 min后获得褐色透明的Au/Pd单原子催化剂溶胶。DFT理论计算表明Au原子在Pd团簇(111)面的吸附能更大,因此Au原子更易于沉积在该面上;由于Au和Pd之间的电荷转移效应,该催化剂催化葡萄糖氧化的活性是单金属Au或Pd团簇催化活性的17~40倍。
2Cu+2OH-+Ni2+ →Cu2O +Ni +H2O
最后将产物在H2气氛中还原得到不同负载量((0.01~0.001) wt%)的Ni1/Cu单原子催化剂。采用类似的方法,他们将Pt和Pd沉积到Cu表面制备了Pt1/Cu、Pd1/Cu单原子催化剂进行对比实验。通过对比不同催化剂在各种温度下的乙醇脱氢生成乙醛反应的活化能可知,Cu纳米颗粒的活化能为(70 ± 5) kJ/mol,而Ni/Cu单原子催化剂的活化能仅为(45 ± 4) kJ/mol,表明在催化剂中Ni单原子的存在会显著降低乙醇脱氢的活化能;DRIFT的结果表明:Ni1/Cu单原子催化剂参与了C—H键裂解,降低了C—H键裂解所需的温度,加快了乙醇的脱氢速率,最终促进了乙醛的生成。X射线吸收精细结构谱(EXAFS)的结果表明:催化剂中存在Ni—Cu、Ni—Ni和孤立的Ni原子,且Ni—Cu的键长小于Ni—Ni键和Cu—Cu键;这是由于孤立的Ni原子和Cu之间的电荷转移影响了Ni—Cu的键长。分散的Ni原子和Cu原子之间的电荷转移作用提高了Cu在高温下的稳定性,降低了C—H键活化能,从而提高了Cu在乙醇脱氢中的催化活性。
Bakr等[48]采用电化学沉积法,通过Au3+还原Ag25纳米团簇核心位置的Ag原子制备出了AuAg24单原子催化剂。相比于直接还原等方法,该方法制得的纳米团簇不含其他杂质。Au原子取代Ag25纳米团簇的核心位置,在不改变其结构的前提下,调控了Ag25团簇的电子结构。同时,Au原子取代后,团簇的稳定性也提高了。
化学还原和电化学沉积法都可以将金属单原子逐步还原并较好地负载在纳米团簇表面。但这些方法的制备过程复杂,同时所制备的单原子催化剂的结构不易调控,且催化剂的耐久性较差,在某些时候其高的催化活性仅能持续10~20 min,难以工业化生产并投入使用。
4 浸渍法制备单原子催化剂
Lang等[40]以RhCl3为催化剂前驱体,以ZnO纳米线为载体,采用浸渍法制备了Rh1/ZnO单原子催化剂。他们先在室温下通过搅拌将ZnO纳米线均匀分散在水溶液中,随后将RhCl3水溶液加入该悬浮液中,经过滤干燥后,在H2/He混合气氛下还原,最终得到了不同负载量(0.3 wt%、0.03 wt%及0.006 wt%)的Rh1/ZnO单原子催化剂。结果表明:ZnO纳米线上Rh的平均直径仅有0.9 nm,且主要是以单个原子的形式负载在ZnO载体上。XPS和XANES光谱表明Rh原子为金属态或略呈负价态,占据了ZnO载体中的O空位;其原因可能是在H2还原的过程中,Rh原子会与邻近的Zn原子发生键合,使Zn失去一个或多个O原子,原本与O结合的电子从Zn原子转移到Rh原子。Rh原子和ZnO之间强的金属-载体相互作用使该催化剂具有高的催化活性和稳定性,且当负载量为0.006 wt%时,Rh1/ZnO催化剂对苯乙烯的选择性达到99%,是传统Wilkinson催化剂RhCl(PPh3)3的10倍以上。另外,通过离心的方式回收催化剂并重复使用4次后,催化剂的活性和选择性仍没有明显降低。
Kim等[41]以H2PtCl6·6H2O为催化剂前驱体,以锑掺杂的氧化锡(ATO)粉体为载体,采用浸渍法制备了Pt1/ATO单原子催化剂。他们先将不同含量的Pt(1 wt%、4 wt%及8 wt%)前驱体溶液与ATO混合,干燥后在H2气氛下分别经100 ℃和400 ℃还原制备Pt1/ATO单原子催化剂。结果表明:100 ℃还原后可得到尺寸为(2.5±0.7) nm的Pt纳米团簇;而当还原温度为400 ℃时,在TEM中观察不到Pt的纳米团簇,但在能谱结果中发现Pt存在;HAADF-STEM图像进一步证实了Pt单原子的存在,且Pt单原子位于SnSb或SnO2表面。密度泛函理论(DFT)的计算结果表明:当SnSb中的Sb位点被Pt替代时,其在热力学上达到最稳定的状态。经过1800次循环后,Pt/ATO仍然保持着较高的催化甲酸氧化反应活性,而商业的Pt/C催化剂则失去催化活性;使用Pt/ATO催化剂作为阳极制作了甲酸燃料电池,该催化剂显示出了高的催化活性、选择性及耐久性。
Narula等[43]以H2PtCl6溶液为催化剂前驱体,以Al2O3为载体,采用浸渍法制备了Pt/θ-Al2O3单原子催化剂。他们先用溶胶凝胶法制得θ-Al2O3载体,再将所制得的θ-Al2O3载体加入到H2PtCl6溶液中,在油浴中浸渍搅拌后加热蒸发,最后在H2中处理制备了Pt/θ-Al2O3单原子催化剂。与Zhang等[17]制备的Pt/FeO x 不同的是,该方法制备的单原子Pt仅吸附在θ-Al2O3的表面。XANES结果表明:在H2的作用下,当催化剂中Pt的负载量为1.0 wt%时,Pt为金属态;而当催化剂的负载量为0.18 wt%时,即使在H2的作用下,Pt依然为氧化态,由此说明负载量为0.18 wt%时的单原子Pt与θ-Al2O3之间存在着强相互作用,使得Pt/θ-Al2O3 单原子催化剂展现出高的催化CO氧化活性,是传统的Pt/θ-Al2O3载体型催化剂的6~18倍。
Zeng等[39]以2-甲基咪唑(C4H6N2)、ZnO和乙酸铁为原料,以H2PtCl6为催化剂前驱体,采用浸渍法制备Pt1@Fe-N-C单原子催化剂。他们先制备Fe-N-C载体,之后将H2PtCl6溶液加入到70 ℃的Fe-N-C溶液中使Pt4+充分吸附,经搅拌、过滤及干燥后,在Ar气氛中热处理制备了负载量约为2.1 wt%的Pt1@Fe-N-C单原子催化剂。研究结果表明:Pt原子均匀分散在载体上,热处理后并没有出现明显的团聚现象(图5a);同时,催化剂中Pt和Fe的价态降低,表明Pt4+离子负载到Fe-N-C上后,Pt和Fe共用两个氧原子,使得其结构发生了一定程度的改变(图5b);为了进一步检测所制备催化剂的耐久性,在饱和H2SO4(0.5 mol·L-1)溶液中分别加载0.3 V和1.1 V的电压进行加速电压循环测试。结果表明:经过10 000次循环后,Pt1@Fe-N-C催化剂的电势仅衰减12 mV,表明其具有良好的耐久性;原因是催化剂中Pt-O2-结构的存在减缓Fenton反应(式(2))的进行。

此外,他们指出羟基自由基(·OH)浓度的降低可以抑制催化剂在最初15 h内发生的表面电化学氧化,在一定程度上提高了活性位点在酸性质子交换膜燃料电池中的稳定性。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
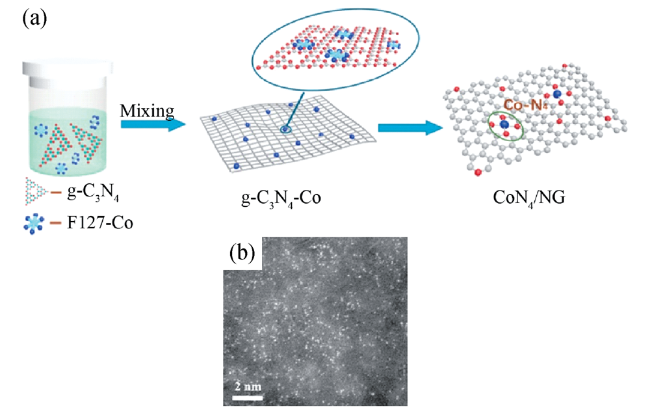
结果表明:表面活性剂F127的加入对Co单原子的分散至关重要,带负电的F127可以吸引正价Co2+离子,使得Co可以均匀地分散在g-C3N4层间;同时F127与周围的氮原子在高温下形成CoN x ,可以有效地防止热处理期间Co金属颗粒的团聚;EXAFS结果表明:在CoN4/氮掺杂石墨烯单原子催化剂中,Co单原子分散在氮掺杂石墨烯上,1个Co原子与4个N原子配位;所制备的CoN4/氮掺杂石墨烯单原子催化剂表现出优异的氧还原反应(ORR)和析氧反应(OER)活性,其半波电位为0.87 V,比20 wt%的Pt/C高20 mV。OER的测试结果表明:CoN4/氮掺杂石墨烯催化剂在10 mA·cm-2下具有380 mV的低过电位,其出色的ORR和OER性能归因于原子分散的CoN4活性位点;以CoN4/氮掺杂石墨烯为催化剂制备的锌-空气电池,经过100 h的循环测试后,其充放电电压间隙仍为0.84 V,没有任何衰减,其能量密度高达671 Wh·kg-1。在柔性固体锌空气电池中实现了28 mW·cm-2的高功率密度和6 h的持续时间,证明了CoN4/氮掺杂石墨烯是一种高效且有前景的单原子催化剂。
与Yang等[42]采用N元素稳定Co催化剂的思路相似,Cheng等[52]以Ni(NO3)2为催化剂前驱体,以微波剥离的石墨烯为载体,以尿素为表面活性剂,制备了负载量约为6.9 wt%的Ni/石墨烯单原子催化剂。结果表明:微波剥离的氧化石墨烯具有高的比表面积(2649 m2·g-1)和小于6 nm的孔结构,浸渍Ni催化剂后,其比表面积和孔结构没有明显变化;微波剥离石墨烯的独特结构有助于Ni单原子的稳定并防止其在高温下的团聚,为单个Ni原子的稳定存在提供了大量锚定点。X射线吸收光谱(XAS)和球差校正透射电子显微镜表明:N稳定的Ni单原子主要被锚定在纳米孔(<6 nm)的边缘。所制备的催化剂在电还原CO2反应中显示出53.6 mA·mg-1的催化活性,在0.59 V的过电位下仍具有92.1%的高选择性;通过DFT计算配位数不同的N与Ni的吉布斯自由能可知,NiN2、NiN2(NH2)、NiN2(NH2)2和NiN3优先在NH3气氛下生成,且N配位Ni单原子催化剂在电还原CO2反应中具有更低的活化能。
总之,浸渍法是制备单原子催化剂的有效方法之一,其工艺简单易行,不需要专门的设备就可以进行规模化生产并满足实际应用。该方法的关键在于调控载体和单原子之间的相互作用力,通过控制催化剂前驱体的类型及其与载体锚定点的相互作用,可以在浸渍过程中将金属单原子沉积在载体上,之后再通过热处理来进一步加强负载的金属原子和载体表面之间的相互作用。但该方法制备的单原子催化剂的负载量普遍较低,这是由于当催化剂尺度减小到原子尺度时,催化剂比表面积增大,表面能增大,单原子易于在制备时发生团聚形成团簇,致使催化剂失活,而低负载量的单原子催化剂难以被应用于实际生产。
5 其他方法制备单原子催化剂
原子层沉积法是一种可以将金属以单原子膜的形式一层一层地镀在基底表面的方法[53],通过简单调整原子层沉积的循环次数,即可达到控制纳米团簇大小、形貌以及质量的目的。Sun等[54]以三甲基(甲基环戊二烯基)铂(MeCpPtMe3)为催化剂前驱体,采用原子层沉积法制备了Pt/石墨烯单原子催化剂,并将其用作甲醇燃料电池的阳极材料。他们采用X光吸收精细结构谱(EXAFS)定性分析了不同Pt含量时单原子催化剂中单原子所占的百分比,结果表明:随着原子层沉积次数(50、100及150)的增多,催化剂中Pt-Pt键逐渐增多,说明逐渐有Pt团簇的产生。当沉积50次时,所制备的Pt/石墨烯催化剂中有着很强的Pt—C及Pt—O键,说明Pt与氧化石墨烯之间的结合很牢固;单原子催化剂Pt—Pt键的键长和块状金属Pt的Pt—Pt键的键长相差很大,说明此时样品中存在着单原子Pt或粒径非常小的Pt团簇。XANES的结果还表明:原子层沉积次数较少时,所制备的催化剂中存在着更多的单原子Pt。相比于传统的Pt/C催化剂,Pt/石墨烯的催化活性提高了10倍,同时催化剂的寿命也延长了。类似地,Lu等[55]用原子层沉积法合成了Pd/石墨烯单原子催化剂。他们先制备出氧化石墨烯,再使Pd前驱体Pd(hfac)2选择性地与氧化石墨烯上的某些位点作用,最后再将配体(hfac)2去掉将单原子Pd固定于石墨烯上。Pd/石墨烯单原子催化剂在1,3-丁二烯加成反应中的催化选择性达到100%,1,3-丁二烯的转化率高达95%;同时该单原子催化剂也表现出良好的耐久性。
Tang等[60]用反奥斯瓦尔德熟化法制备了负载在Hollandite型MnO2(HMO)上的AgAOR/HMO单原子催化剂,同时与采用浸渍法制备的AgIMP/HMO单原子催化剂的结构及催化性能进行了对比。XANES的结果表明:通过浸渍法制备的AgIMP/HMO的吸收峰在单质Ag的吸收峰之前,说明此时的d轨道未被填满,这可能是d轨道的电子跃迁导致的;AgAOR/HMO单原子d轨道的电子跃迁强于AgIMP/HMO单原子催化剂,表明该催化剂中金属与载体之间的相互作用更强。甲醛催化氧化实验结果表明:AgAOR/HMO单原子催化剂的活化能为91 kJ·mol-1,其高的催化活性可归因于金属与载体之间的相互作用。
Gu等[50]先用物理气相沉积法制得ZnO纳米线,随后在室温下将一定量的HAuCl4或H2PtCl4溶液加入到ZnO纳米线的水溶液中,搅拌静置一段时间后,过滤、洗涤并干燥后,在390 ℃下的甲醇中得到了负载量仅为0.0125 wt%的Pt/ZnO或Au/ZnO单原子催化剂。单原子的存在为甲醇蒸气重整过程提供了单一、稳定的活性位点,降低了反应势垒,改变了反应路径,其催化活性是ZnO的1000倍。
采用上述方法制备的单原子催化剂都表现出了很好的催化性能,这些方法具有沉积均匀性和重复性好等优点。但在实际应用中仍存在着工艺复杂、设备价格昂贵及催化剂负载量低的问题,严重限制了这些方法在实际情况下的应用。
6 结论与展望
常见的单原子催化剂制备方法有共沉淀法、化学还原法、电化学沉积法和浸渍法。共沉淀法是制备单原子催化剂较经济的方法,可以使不同的物相均匀分布在载体上,但利用该方法制备单原子催化剂的影响因素较多。化学还原及电化学沉积法制备的单原子催化剂产物催化活性好,可以长期存放,但该方法的制备过程复杂,且所制备的单原子催化剂的结构不易调控。浸渍法是制备单原子催化剂工艺简单且有效的方法,其对设备的要求低,是较为理想的制备单原子方法。但采用上述三种方法制备的单原子催化剂普遍存在着负载量偏低的缺点,这是因为单原子催化剂的粒径较小使其表面自由能较高,导致负载量较大时单原子催化剂易于团聚,从而限制了单原子催化剂的应用。
总之,单原子催化剂具有催化效率高、稳定性好及利用率高等优点,在催化制氢、催化氧化及光催化领域有着广阔的应用前景,其出色的催化性能可归因于单个金属单原子的存在。此外,金属单原子与载体之间的强相互作用对单原子催化剂的催化活性有着显著的影响。虽然目前单原子催化剂的研究报道已有很多,但仍存在以下问题:1)单原子催化剂的金属负载量低;2)目前的研究仍只处于实验室研究阶段,鲜有用于工业生产。
基于以上挑战和问题,我们认为今后一段时期单原子催化剂的主要研究方向如下:
(1)开发新的合成方法,提高金属单原子催化剂的负载量,制备具有高催化性能和高负载量的单原子催化剂,为各个领域的催化反应做出贡献;
(2)进一步深入研究单原子催化剂的制备理论和催化机理,总结最优参数和相关规律,设计可用于工业生产的单原子催化剂,推动催化工业的发展进步;
(3)采用具有一定强度的高孔隙率和比表面积的三维多孔材料替代不易回收且易团聚的纳米线、纳米片作为载体制备单原子催化剂,进一步提高其耐久性,并使其更易于被回收循环利用。